

Věstník MŽP ČR, částka 1/2017
Certifikovaná metodika
(Nmet)
Metodika pasivního vzorkování perfluoroktansulfonátu PFOS, PFOA a vybraných farmak ve vodním prostředí
Masarykova univerzita Přírodovědecká fakulta
Centrum pro výzkum toxických látek v prostředí RECETOX
Metodika je výsledkem řešení projektu „Emergentní polutanty ve složkách životního prostředí“ (TB030MZP001) podpořeného TA ČR v rámci programu BETA.
Řešitelský tým projektu:
doc. Ing. Branislav Vrana, Ph.D.
prof. RNDr. Jana Klánová, Ph.D.
doc. RNDr. Pavel Čupr, Ph.D.
RNDr. Roman Prokeš, Ph.D.
RNDr. Jana Borůvková, Ph.D.
Ing. Jitka Bečanová, Ph.D.
Foppe Smedes
doc. Mgr. Klára Hilscherová, Ph.D.
Mgr. Jiří Novák, Ph.D.
Mgr. RNDr. Michal Bittner, Ph.D.
Mgr. Ondřej Sáňka, Ph.D.
Ing. Šimon Vojta, Ph.D.
RNDr. Ondřej Mikeš, Ph.D.
Mgr. Anežka Nečasová
Mgr. Katarína Bányiová
Ing. Kateřina Šebková, Ph.D.
Mgr. Jakub Urík
Ing. Barbora Feixová
Výše uvedení členové týmu jsou z Centra pro výzkum toxických látek v prostředí RECETOX, Přírodovědecké fakulty MU - tedy z řešitelského pracoviště projektu.
Citace: Vrana B. a kol..: Metodika pasivního vzorkování perfluoroktansulfonátu PFOS, PFOA, PFC vybraných farmak ve vodním prostředí - certifikovaná metodika (Nmet). Masarykova univerzita, RECETOX. RECETOX REPORT No. 598. Listopad 2016.
5 Princip pasivního vzorkování
6 Pasivní vzorkování polárních organických látek - adsorpční pasivní vzorkovače
7.1 Příprava vzorkovačů
7.1.1 Potřebné množství vzorků
7.1.2 Uskladnění vzorkovačů
7.2 Výběr odběrového místa
7.3 Pasivní vzorkování v terénu
7.3.1 Instalace pasivních vzorkovačů v terénu
7.3.2 Vyjímání vzorkovačů
7.4 Odeslání vzorků k analýze
7.5 Analytické stanovení perfluorovaných látek a vybraných léčiv v extraktech z pasivních vzorkovačů
7.5.1 Extrakce vzorkovačů
7.5.2 Instrumentální analýza
7.6 Kalibrační parametry vzorkovače
7.7 Interpretace výsledků měření
7.7.1 Příklad stanovení rozpuštěné koncentrace perfluorovaných látek v povrchové vodě pomocí pasivního vzorkování
7.8 Zajištění a řízení kvality (QA/QC)
7.8.1 Činidlo pro slepé stanovení (reagent blank)
7.8.2 Laboratorní kontrola
7.8.3 Terénní kontrola
7.8.4 Zařízení pro určení výtěžnosti (recovery spike)
7.8.5 Použití kontrolních a slepých vzorků
7.8.6 Řízení kvality v přípravě vzorků na toxikologickou analýzu
7.9 Zpráva o zkoušce
8 Popis novosti a uplatnění metodiky
10 Protokol o odběru vzorků (vzor)
11 Stanovení distribučních koeficientů sledovaných látek v soustavě adsorbent-voda (KSW)
11.1 Princip metody
11.2 Stanovení KSW
12.1 Princip metody
12.2 Stanovení difúzních koeficientů látek v agarozovém hydrogelu
12.3 Postup stanovení
13 Kalibrace pasivního vzorkovače v průtokovém systému
14 Příklady podmínek instrumentální analýzy
14.1 Perfluorované látky
14.2 Farmaka
2 Seznam zkratek a symbolů
|
A |
plocha vzorkovače, kterou difunduje analyt |
|
APV |
adsorpční pasivní vzorkovač |
|
Cfree |
koncentrace volně rozpuštěné látky |
|
ChA |
chemická aktivita látky |
|
D |
difúzní koeficient |
|
DCM |
dichlormethan |
|
DEQ |
stupeň dosažení rovnováhy v systému vzorkovač-voda pro sledovanou látku |
|
DGT |
vzorkovač s difúzním gradientem v tenké vrstvě |
|
dMeFOSA |
d-N-methyl-perfluorooktansulfonamid |
|
dMeFOSE |
d-N-methyl perfluorooktan sulfonamidoethanol |
|
EEO |
elektroendoosmóza; míra mobility tekutiny v gelu |
|
ESI |
ionizace elektrosprejem |
|
GC-MS |
plynová chromatografie s hmotnostní spektrometrií |
|
ji |
difúzní tok látky i-tou fází |
|
ke |
eliminační rychlostní konstanta prvního řádu |
|
ki |
koeficient přestupu látky i-tou fázi |
|
Kmw |
rozdělovací koeficient membrána-voda |
|
Kow |
rozdělovací koeficient oktanol-voda |
|
Kpw |
rozdělovací koeficient vzorkovač-voda; adsorbent-voda |
|
LOD |
mez detekce metody |
|
LOQ |
mez stanovení metody |
|
mp |
hmotnost sorpční fáze vzorkovače |
|
M8PFOA |
perfluoro-n-[13C8]oktanová kyselina |
|
M8PFOS |
perfluoro-[13C8]oktansulfonát |
|
MRM |
multiplexní analýza (multiple reaction monitoring) |
|
NEK |
norma environmentální kvality |
|
PFC |
perfluorované látky (perfluorinated compounds) |
|
POP |
persistentní organické látky |
|
PPCPs |
léčiva a látky osobní péče (pharmaceutical and personal care products) |
|
PRC |
provozní referenční látky (performance reference compounds) |
|
Q |
průtok vody v systému; objem vody vyměněný za jednotku času |
|
R |
univerzální plynová konstanta |
|
Re |
Reynoldsovo číslo |
|
RPV |
rozdělovací pasivní vzorkovač |
|
RS |
rychlost vzorkování (sampling rate) |
|
Sc |
Schmidtovo číslo |
|
Sh |
Sherwoodovo číslo |
|
SPE |
extrakce na tuhé fázi |
|
SPME |
mikroextrakce na tuhé fázi |
|
Sw |
rozpustnost látky ve vodě |
|
TWA |
time-weighted average; časově vážená průměrná koncentrace |
|
VRS |
Vodní Rámcová Směrnice 2000/60/ES |
|
WBL |
mezní difúzní vrstva vody (water boundary layer) |
Klíčová slova
Pasivní vzorkovací zařízení, odběr vzorků v povrchových vodách, perfluorované organické látky, farmaka, volně rozpuštěná koncentrace
Terminologie
V metodice je dále použita terminologie definována v ČSN EN ISO 5667-23 Jakost vod - Odběr vzorků - Část 23: Návod pro pasivní odběr vzorků v povrchových vodách [1].
3 Cíl metodiky
Metodika popisuje metodu a zařízení pro účinné pasivní vzorkování perfluorovaných látek a jiných organických látek polárního charakteru z vodního prostředí, které umožňuje kvantifikaci volně rozpuštěné koncentrace těchto látek ve vodním sloupci v útvarech povrchových vod.
Pasivní vzorkování je monitorovací nástroj, kterého lze vhodně využít pro monitorování organických látek ve vodě. Je navržen tak, aby poskytnul informaci o volně rozpuštěných koncentracích sledovaných látek, které jsou v mnoha případech nejvhodnějším parametrem, umožňujícím vysvětlení expozice vodních organizmů a toxických účinků látek na vodní živočichy. Časově integrační povaha odběru vzorků v kombinaci s použitím vzorkovacího media (sorbentu) s dobře definovanými a konstantními vlastnostmi umožňuje, aby se minimalizovala variabilita monitorovacích dat v důsledku vzorkování. Pasivní vzorkování je vhodným nástrojem k monitorování stopových organických znečišťujících látek za účelem hodnocení jejich časových a prostorových trendů v povrchových vodách.
Širšímu využití pasivního vzorkování polárních látek v praxi zatím částečně brání, že na rozdíl od bodových odběrů vzorků vody, v současnosti komerčně dostupné pasivní vzorkovače polárních látek typu POCIS [2] poskytují pouze semikvantitativní informaci o koncentracích látek ve vzorkované vodě [3]. Je to kvůli použití polyetersulfonové difůzní membrány, která částečně sorbuje sledované sloučeniny, a také nedostatečně tlumí vliv rychlosti proudění vody na vzorkovací rychlost zařízení [3]. Navzdory tomuto omezení adsorpční pasivní vzorkovače mohou poskytnout reprezentativnější informaci o znečištění vod než bodové odběry, zejména v případech, kdy variabilita koncentrace sledované látky ve vodě je vyšší, než je nejistota spojená s pasivním odběrem [4]. Cílem předložené metodiky je poskytnout pasivní vzorkovač, který umožňuje stanovení perfluorovaných látek a vybraných farmak ve vodním prostředí s definovanou nejistotou měření.
4 Popis metodiky
Předložená metodika představuje nový pasivní vzorkovač založený na adsorpci sledovaných látek (perfluorované látky a vybraná farmaka) z vodního prostředí do vhodného adsorbentu. Přestup látky z vody do vzorkovače je kontrolován difúzí ve vrstvě agarozového hydrogelu. Přestup látky lze dobře kvantifikovat pomocí difúzních koeficientů sledovaných látek v hydrogelu, které byly stanoveny jako součást metodiky. Volbou vhodné tloušťky hydrogelu lze kontrolovat přestup látky, a tím i vzorkovací rychlost látek do vzorkovacího zařízení. Vzorkovací rychlosti navrženého vzorkovače jsou robustní a mohou být aplikovány jako kalibrační parametr vzorkovače, který umožňuje v principu přepočítat množství sledované látky ve vzorkovači na časově vážený průměr koncentrace ve vodní fázi ve vzorkovaném vodním útvaru.
Součástí metodiky je
-
návrh adsorbentu vhodného pro konstrukci pasivního vzorkovače perfluorovaných látek a vybraných farmak ve vodním prostředí
-
stanovení kalibračních parametrů pasivního vzorkovače - difúzních koeficientů perfluorovaných látek v agarozovém hydrogelu (a souvisejících laboratorních vzorkovacích rychlostí vzorkovače pro sledované látky)
-
návrh zařízení pro pasivní vzorkování perfluorovaných látek z povrchových vod
-
metodický postup extrakce a úpravy vzorku pasivního vzorkovače před analýzou perfluorovaných látek LC-MS
-
metodický postup interpretace dat - algoritmus výpočtu koncentrace perfluorovaných látek ve vodě z dat z pasivního vzorkování.
Metoda je určena pro aplikaci v povrchové vodě. Je možné ji adaptovat i k pasivnímu odběru pitné a podzemní vody. Povrchově aktivní látky, emulgátory, vyšší koncentrace polárních rozpouštědel nebo zvýšená hodnota vodivosti vody mohou ovlivnit akumulaci sledovaných látek do vzorkovače. Přítomnost druhé fáze ve vodě (např. ropné látky, emulgované tuky a vosky) může rušivě působit na proces odběru vzorků. Metoda byla testována v rozsahu pH vody 6-8. Extrémní hodnoty pH mohou ovlivňovat hodnoty vzorkovacích rychlostí a distribučních koeficientů látek, zejména pro látky, které ve vodě disociují (např. farmaka). Metoda nebyla ověřena pro použití v komunální a průmyslové odpadní vodě.
5 Princip pasivního vzorkování
Pasivní vzorkování je založeno na použití in situ zařízení, které akumuluje kontaminanty z vody. Přestup kontaminantu z prostředí do vzorkovače je samovolný difúzní proces, který je hnán rozdílem chemických aktivit monitorované látky mezi vzorkovaným médiem a sorpční fází vzorkovače (Obrázek 1).
Akumulace látky ve vzorkovači probíhá až do ustálení termodynamické rovnováhy (resp. ustáleného stavu v otevřených dynamických systémech, jakými jsou např. řeky) mezi vzorkovačem a vodou, anebo dokud se proces vzorkování nepřeruší. Doba expozice vzorkovačů je obvykle několik dní až týdnů. Akumulované kontaminanty se následně extrahují a v extraktu se stanoví jejich koncentrace. Pokud jsou vzorkovače kalibrované, je možné z množství látky ve vzorkovači vypočítat koncentraci látky rozpuštěné ve vzorkovaném médiu, např. ve vodě. Pasivní vzorkování je často integrační, tj. získaný vzorek reprezentuje koncentraci látky ve vzorkovaném médiu za určité časové období. Velmi důležitým aspektem pasivního vzorkování je možnost vyjádřit množství látky ve vzorkovači v rovnováze se vzorkovaným médiem formou chemické aktivity, která je mírou hnací síly pro samovolný přestup látky mezi různými složkami životního prostředí [5].
V počáteční fázi expozice vzorkovače se látky z vody akumulují do vzorkovače rychlostí, která je přímo úměrná jejich koncentraci ve vodní fázi (lineární fáze vzorkování; Obrázek 2). V lineární fázi odběru vzorek reprezentuje časově vážený průměr koncentrace látky ve vzorkovaném médiu (vodě) a vzorkování se označuje jako časově integrativní. Po delší době expozice vzorkovče je dosaženo distribuční rovnováhy látky mezi vzorkovačem a vzorkovaným médiem. Tato fáze odběru se nazývá rovnovážné pasivní vzorkování. Díky vysoké sorpční kapacitě (Ksw×ms) a integračnímu charakteru pasivních vzorkovačů je možné monitorovat látky, které se nacházejí rozpuštěné ve vodě v extrémně nízkých koncentracích (řádově pg l-1).
Konvenční metody vzorkování vody, založené na bodových odběrech, neumožňují stanovení takových nízkých koncentrací, ačkoli normy environmentální kvality (NEK) určené Vodní rámcovou směrnicí [6- 8] vyžadují monitorovat některé znečišťující látky ve vodě metodami, které mají mez stanovení na úrovni ng l-1 i níže. V odborné literatuře je dostupných několik přehledových prací, které popisují design, kalibrační postupy, pracovní charakteristiky a příklady aplikace různých pasivních vzorkovačů na monitorování znečišťujících látek ve vodním prostředí [9-21]. Booij a kol. [22] sumarizovali potenciál využití různých pasivních vzorkovačů pro monitorování látek regulovaných Vodní rámcovou směrnicí [6] a jinými směrnicemi a úmluvami (OSPAR [23]; U.S. Clean Water Act atd.). Vrana a kol. [18] vypracovali pro asociaci laboratoří NORMAN stanovisko, který uvádí přehled použitelnosti pasivního vzorkování pro monitorování emergentních (dosud neregulovaných) znečišťujících látek ve vodním prostředí. Další aktuální poziční dokument asociace NORMAN o pasivním vzorkování byl publikován mezinárodní skupinou expertů, na základě diskusí na specializovaném workshopu, který se konal v listopadu 2014 v Lyonu [24]. Dokument identifikuje konkrétní aktivity, které je nutné vykonat, aby pasivní vzorkování mohlo být v budoucnu využíváno v rutinním monitoringu vodního prostředí za účelem posouzení rizik, managementu kontaminantů a k hodnocení jejich trendů v životním prostředí. Užitečným zdrojem informací o principech a aplikacích pasivního vzorkování ve vodním prostředí je přehledová monografie o pasivních vzorkovacích technikách pro monitorování životního prostředí [25].
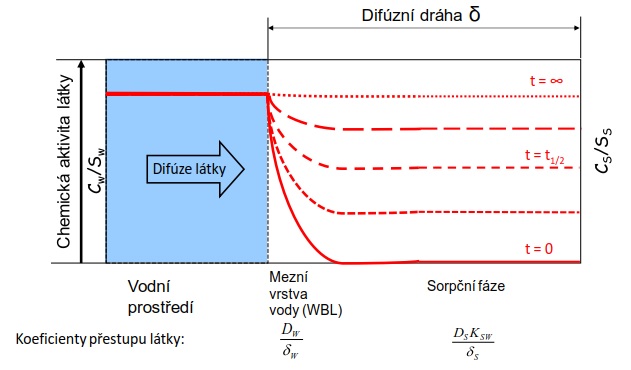
Obrázek 1. Funkční princip pasivního vzorkovače, který ukazuje koncentrační profil látky po dobu difúze a akumulace z vodního prostředí do sorbentu (sorpční fáze) v čase t. Vysoká afinita látky k sorpční fázi je hnací silou difúze molekul sledované látky do vzorkovače, dokud nedojde k vyrovnání chemické aktivity látky v obou médiích, tj. k ustálení termodynamické rovnováhy.
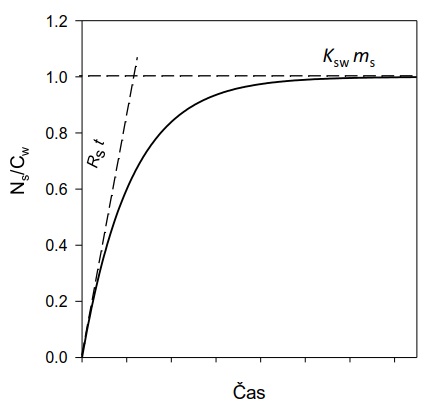
Obrázek 2. Efektivní objem vody extrahovaný vzorkovačem (Ns/Cw) jako funkce času. Pro dlouhé expoziční doby je extrahovaný objem omezen sorpční kapacitou vzorkovače (Ksw×ms) a pro krátké expoziční časy součinem vzorkovací rychlosti a doby expozice. Přibližné modely, které platí pro lineární část akumulace (krátká doba expozice) a rovnovážné vzorkování (dlouhá doba expozice) jsou znázorněny čárkovanými čarami. Upraveno podle [26].
6 Pasivní vzorkování polárních organických látek - adsorpční pasivní vzorkovače
Základem adsorpčních pasivních vzorkovačů (APV) je jeden nebo více typů sorbentu, oddělených od vnějšího prostředí obvykle membránou a případně jinou difúzní vrstvou. Ve vzorkovači dochází k difúzi látek přes porézní membránu a difúzní vrstvu a k následné sorpci na selektivní adsorpční materiály. Důvodem akumulace je vysoká afinita rozpuštěné látky k sorbentu, respektive výrazný rozdíl v chemické aktivitě látky volně rozpuštěné ve vodě a stejné látky sorbované ve vzorkovači. Sorbent může vázat sledované látky interakcemi různeho charakteru (např. hydrofobní, iont-iont, dipól-dipól, vodíková vazba, van der Waalsovy interakce). V případě použití sorbentu na bázi iontoměniče, budou převažovat iontové interakce a vzorkovač bude zachycovat látky s příslušnou ionizovatelností. Naopak málo polární neutrální látky sorbovány nebudou. Proto je pomocí APV, narozdíl od rozdělovacích pasivních vzorkovačů (RPV), možné vzorkovat i vysoce polární látky. Zároveň při použití polymerů, které obsahují ve své struktuře hydrofilní i lipofilní funkční skupiny, (např. sorbent Oasis HLB®), jsou APV schopny vzorkovat i nepolární látky, čímž se stávají širokospektrálním vzorkovacím nástrojem. K sorpci látek dochází po celou dobu expozice, až do vyčerpání sorpční kapacity sorbentu. Jelikož však na sorbentu dochází k různým typům interakcí, kapacita je zpravidla odlišná pro různé látky. Kapacita sorbentu je závislá na použitém množství adsorbentu (hmotnost sorpční fáze je označována ms) a na distribučním koeficientu sledované látky mezi adsorbentem a vodním roztokem (Ksw) a je dána jejich součinem (Ksw×ms). Obvykle je kapacita sorbentu dostatečně vysoká na to, aby se sorbent choval jako "nekonečný příjemce" a množství akumulované látky se zvyšovalo lineárně, v závislosti na vnější koncentraci po celou dobu expozice vzorkovače (několik dnů až týdnů). Pokud je kapacita sorbentu nedostatečná, je vhodné použít jiný typ sorbentu, přiměřený vlastnostem sledovaných látek. Pomocí difúzních koeficientů (D) nebo kalibrací zjištěných vzorkovacích rychlostí (Rs) pro danou látku je možné vypočítat koncentraci volně rozpuštěné látky ve vodě (Cfree), respektive její časově vážený průměr za celou dobu vzorkování (TWA). Tato koncentrace se může, zejména pro hydrofobní látky, lišit od celkové koncentrace ve vodě, protože ta zahrnuje i látky navázané na různé částice dispergované ve vodním sloupci. Cfree je však vhodným parametrem pro popis distribuce látky mezi jednotlivými složkami životního prostředí, jakož i vhodným odhadem biodostupné frakce látky ve vodě. Na rozdíl od rozdělovacích procesů probíhajících v RPV se sorpce a desorpce látek v APV neřídí izotropní kinetikou prvního řádu. Proto je použití provozních referenčních látek (PRC) [27] při pasivním vzorkování polárních látek značně omezené. Přestože existují snahy najít potenciální PRC látky vhodné pro adsorpční pasivní vzorkovače, jsou většinou použitelné pouze pro úzký okruh podobných látek a pro skupinu perfluorovaných látek vhodné nejsou [28-30].
|
|
Zdroje dalších informací: Adsorpční pasivní vzorkování Přestože existuje velké množství publikací o vývoji, kalibraci a použití adsorpčních pasivních vzorkovačů [3,31-34], není k dispozici mnoho oficiálních příruček. Velmi obecná technická ISO norma byla publikována v roce 2011 [35] a příručku, jak používat vzorkovač POCIS lze nalézt na webových stránkách USGS [36], na Wikipedii [37] a v češtině je dostupná také metodika, publikována Grabicem a kol [38]. Tyto zdroje informací obsahují praktické rady na použití pasivního odběrového zařízení, ale část interpretace dat není dostačující, neboť chybí jasné deterministické metody pro odhad vzorkovaného objemu vody pasivním odběrem v terénu. |

7 Sledované sloučeniny
Tabulka 1. Sledované sloučeniny: Perfluorované látky
|
Látka |
Zkratka |
CAS |
Vzorec |
M (g mol-1) |
log Kow |
|
Kyselina perfluorpentanová |
PFPA |
422-64-0 |
CF3(CF2)3COOH |
164,0 |
- |
|
Kyselina perfluorhexanová |
PFHxA |
307-24-4 |
CF3(CF2)4COOH |
314,1 |
4,06 |
|
Kyselina perfluorheptanová |
PFHpA |
1546-95-8 |
CF3(CF2)5COOH |
346,1 |
4,67 |
|
Kyselina perfluoroktanová |
PFOA |
335-67-1 |
CF3(CF2)6COOH |
414,1 |
5,30 |
|
Kyselina perfluornonanová |
PFNA |
375-95-1 |
CF3(CF2)7COOH |
464,1 |
5,92 |
|
Kyselina perfluordekanová |
PFDA |
335-76-2 |
CF3(CF2)8COOH |
514,1 |
6,50 |
|
Kyselina perfluordodekanová |
PFDoDA |
307-55-1 |
CF3(CF2)10COOH |
614,1 |
7,77 |
|
Kyselina perfluortridekanová |
PFTrDA |
72629-94-8 |
CF3(CF2)11COOH |
664,1 |
8,25 |
|
Kyselina perfluorbutansulfonová |
PFBS |
59933-66-3 |
CF3(CF2)3SO3H |
300,1 |
3,90 |
|
Kyselinaperfluorhexansulfonová |
PFHxS |
355-46-4 |
CF3(CF2)5SO3H |
400,1 |
5,17 |
|
Kyselina perfluorktansulfonová |
PFOS |
45298-90-6 |
CF3(CF2)7SO3H |
500,1 |
6,43 |
|
Perfluorooktansulfonamid |
FOSA |
754-91-6 |
CF3(CF2)7SO2NH2 |
499,1 |
5,62 |
Tabulka 2. Sledované sloučeniny: Farmaka
|
Látka |
CAS |
Vzorec |
M (g mol-1) |
log Kow |
pKa |
|
Karbamazepin |
298-46-4 |
C15H12N2O |
236,3 |
2,45 |
13,9 |
|
Sulfamethoxazol |
723-46-6 |
C10H11N3O3S |
253,3 |
0,89 |
1,60 5,70 |
|
Diklofenak |
15307-86-5 |
C14H11Cl2NO2 |
296,1 |
4,51 |
4,15 |
7.1 Příprava vzorkovačů
Předložená metodika představuje nový pasivní vzorkovač založený na adsorpci sledovaných látek (perfluorované látky a vybraná farmaka) z vodního prostředí do vhodného adsorbentu. Přestup látky z vody do vzorkovače je kontrolován difúzí ve vrstvě agarozového hydrogelu.
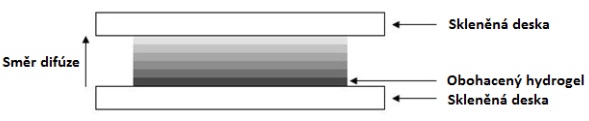
Pro výrobu vzorkovačů na bázi agarozového hydrogelu se do 100 ml destilované vody naváží 1,5 g agarózy a směs je za neustálého míchání na magnetické míchačce zahřívána tak, aby nedosáhla bod varu, ale aby došlo k rozpuštění agarózy. To je indikováno zprůhledněním roztoku. Aby nedocházelo k odparu vody, je nutné kádinku s roztokem přikrýt skleněnou deskou nebo hodinovým sklem (Obrázek 3 vlevo).
K přípravě sorpční vrstvy hydrogelu s dispergovaným sorbentem (Obrázek 4) je po vyčeření roztoku k 100 ml horkého roztoku 1,5% agarózy přidáno 5 g předčištěného sorbentu Oasis HLB® (sorbent je přečištěn methanolem). V závislosti na cílových analytech je možné použít i jiný sorbent. Například v případě kyselin a perfluorovaných látek s krátkým řetězcem je vhodné použít např. sorbent Oasis WAX® nebo jiný slabý anex. Pro výrobu vlastní tenké vrstvy gelu s dispergovaným adsorbentem je nutné vlít připravený roztok/suspenzi mezi dvě skleněné tabule o velikosti 10×10 cm. K dávkování roztoku je vhodné využít automatických pipet s přesným dávkováním roztoku. V závisloti na požadované tloušťce vrstvy gelu (typická tloušťka je 0,5 cm) se mezi vrstvy umístí teflonový mezerník a celý systém se upevní kancelářskými svorkami (Obrázek 3 vpravo). Vychladnutím při laboratorní teplotě nebo pod proudem studené vody pak vznikne tuhý plátek hydrogelu, který může být následně nařezán ostrým nožem do požadovaného tvaru a rozměrů. Vyrobené pláty hydrogelu je možné skladovat v destilované vodě v chladničce při 4°C. Nicméně, kvůli zamezení kontaminace připravených plátů gelu, je vhodné je připravovat v co nejkratším možném čase před samotným vzorkováním, případně skladovat v směsi roztoku methanol/voda (např. 50% v/v).

Obrázek 3. Příprava filmů agarozového hydrogelu o definované tloušťce
K přípravě difúzní vrstvy hydrogelu je použita komerčně dostupná nylonová síť s velikostí ok 2 mm. Tuto síť je nutné před přípravou vlastního vzorkovače rozstříhat na rozměry větší než 10×10 cm. Do jednotlivých kusů sítí je nutné vyrazit díry kopírující umístění děr v kovových prstenech držáku vzorkovače (Obrázek 4). Následně je potřeba sítě pročistit v methanolu ultrazvukovou lázní po dobu 15 minut a dále skladovat v methanolu v chladničce. Před použitím je vhodné sítě vysušit na vzduchu a následně při vytváření gelu vložit mezi teflonové mezerníky. Další postup se shoduje s přípravou sorpční vrstvy hydrogelu.
Připravené pláty gelu jsou ručně uspořádány mezi kovové prsteny spolu s teflonovými mezerníky, a ty pak upevněny pomocí tří šroubů (Obrázek 4). Hotové vzorkovače jsou skladovány ve vzorkovnici s roztokem azidu sodného v destilované vodě (koncentrace azidu sodného cca 0,1%) na potlačení případného mikrobiálního růstu.

Obrázek 4. Schéma konstrukce pasivního vzorkovače založeného na principu adsorpce polárních látek z vodního prostředí do adsorbentu dispergovaného v agarozovém hydrogelu. Přestup látky z vody do vzorkovače je kontrolován difúzí ve vrstvě čistého agarozového hydrogelu.

Obrázek 5. Sestavený pasivní vzorkovač na principu adsorpce polárních látek z vody do adsorbentu dispergovaného v agarozovém hydrogelu.
7.1.1 Potřebné množství vzorků
Každá šarže se skládá ze vzorkovačů pro všechna plánovaná místa odběru v rámci jedné vzorkovací kampaně. Tento počet se zvyšuje o minimálně tři laboratorní kontrolní vzorky (7.8.2), které se neexponují v terénu, ale jsou využívány s cílem zjistit možnou kontaminaci vzorkovačů během přípravy, a též k výpočtu meze stanovení. Dále se do vzorkovacího plánu doporučuje zahrnout cca 20% terénních kontrol (7.8.3). Terénní kontroly jsou vzorkovače, které nejsou nasazeny v terénu ve vodě, ale exponují se na vzduchu během nasazení a vyjmutí exponovaných vzorkovačů. Počet potřebných vzorkovačů (n), který je potřebný pro konkrétní terénní kampaň, se vypočítá z počtu míst odběru vzorků (nmíst):
n = [1.2nmíst + MAX(nmíst/10,3)] (Rovnice 1)
|
|
Pozor! Kvůli opakovatelnosti a vzájemné porovnatelnosti vzorků je důležité, aby série vzorkovačů určená pro jednu terénní kampaň byla zhotovena z materiálu homogenního složení a v jedné šarži. |

7.1.2 Uskladnění vzorkovačů
Vzorkovače jsou baleny do hliníkové fólie a pak do vzduchotěsných plastových sáčků se zipem částečně naplněných destilovanou vodou. Kvůli zabránění mikrobiální aktivity může být do vody přidán biocid (např. 1% azid sodný, nebo jiná látka, která neinterferuje s chemickou analýzou sledovaných látek).
Sáčky pak mohou být ještě umístěny do vzduchotěsných plastových nádob s víkem. Víka se vzduchotěsně uzavřou a nádoby se skladují při 4°C. Vzorkovače by neměly být v průběhu skladování a transportu zmraženy, aby nedošlo ke změně difúzních a sorpčních vlastností hydrogelových vrstev a jejich popraskání při zmrazování a opětovném rozmrazování. Vzorkovnice by měly být uchovávány v temnu, aby se zabránilo fotolýze sledovaných látek.
7.2 Výběr odběrového místa
Před zahájením studie s pasivními vzorkovači je potřebné vhodně zvolit odběrová místa pro jejich umístění. Výběr místa závisí na účelu, za kterým jsou znečišťující látky monitorovány. Pokud má být sledován stav znečištění stopovými látkami ve vodním útvaru, vzorkovače by měly být umístěny mimo směšovací zóny toků s bodovými zdroji znečištění, jako jsou například vypouštěné odpadní vody z průmyslových a komunálních zdrojů, znečištěné přítoky, kontaminované lokality apod. Je užitečné získat informace o míře turbulence vodního toku, průtoku, teplotě, vodivosti, pH, hloubce vody, stratifikaci, potenciálu pro tvorbu biofilmů a zákalu v místě expozice vzorkovačů. Je potřeba zvážit možnost, zda sledované sloučeniny mohou být podrobeny fotolýze slunečním světlem, a pokud existuje takové riziko, zda vzorkovací zařízení a expoziční podmínky (např. zákal vody, přirozené stínění), bude dostatečně chránit vzorkovače před slunečním zářením. Pokud tomu tak není, může být potřebné realizovat další opatření, aby se zabránilo fotolytickému rozkladu sledovaných látek. Je potřeba i minimalizovat riziko, že budou po dobu expozice vzorkovače mechanicky poškozeny, ukradeny, nebo úmyslně vytaženy z vody. Je vhodné také posoudit, zda je přístup na místo odběru vzorků bezpečný, a zda lze na místě odběru pracovat bez toho, aby bylo zvýšeno riziko kontaminace vzorků při manipulaci.
7.3 Pasivní vzorkování v terénu
Obecná pravidla pro rozmisťování, expozici a vyjímání pasivních vzorkovačů v terénu jsou popsána v normě ČSN EN ISO 5667-23 [1]. Pasivní vzorkovače mohou být nasazeny ve vodě s použitím různých vzorkovacích zařízení. Účelem odběrového zařízení je udržet vzorkovače v požadované poloze ve vodním sloupci a chránit je před poškozením proudem vody nebo předměty unášenými proudem, povětrnostními podmínkami, lodní dopravou nebo vodními organismy [39]. Na rozdíl od rozdělovacích pasivních vzorkovačů nepolárních látek, zhotovených z pružných elastomerů, adsorpční pasivní vzorkovací zařízení jsou zpravidla vyrobeny z materiálů, které jsou křehčí, a může snadněji dojít k mechanickému poškození. To je třeba zvážit při volbě odběrového zařízení, které by mělo umožnit volný pohyb vody kolem vzorkovače, ale současně minimalizovat riziko poškození vzorkovačů silnými vířivými proudy, plovoucími nečistotami atd. Některá zařízení, např. klece z perforovaných ocelových plátů, také poskytují zastínění vzorkovačů, čímž snižují riziko fotodegradace sledovaných látek. Pasivní vzorkovače mohou často být snadno nasazeny za použití levných odběrových zařízení, jako jsou mříže z nerezové oceli (viz. Obrázek 6 vlevo). Robustnější držáky jsou potřebné při vzorkování pod silným proudem vody, kdy zařízení musí chránit vzorkovače před mechanickým poškozením (viz. Obrázek 6 vpravo). Při použití vzorkovačů v odpadní vodě musí být všechny komponenty vyrobeny z materiálů odolných vůči korozi kovů (např. nerezová ocel 316L nebo titanová slitina). Příklady různých vzorkovacích zařízení lze nalézt i v literatuře [36,39,40].

Obrázek 6. Příklady různých způsobů instalace pasivních vzorkovačů v terénu
Vzorkovací zařízení by měla být vybavena upevňovacím okem, které umožní zavěšení vzorkovačů na bóji, na mostním tělese nebo na mole. Uzly mohou být zajištěny stahovacími pásky. Příklad instalace vzorkovače je znázorněn na obrázku 6.
7.3.1 Instalace pasivních vzorkovačů v terénu
Při rozmísťování vzorkovačů v terénu platí všeobecná pravidla uvedená v normě ČSN EN ISO 5667-23 Jakost vod - Odběr vzorků - Část 23: Návod pro pasivní odběr vzorků v povrchových vodách [1].
7.3.1.1 Materiál potřebný na instalaci pasivních vzorkovačů
-
Vzorkovací zařízení - držák vzorkovače
-
Vzorkovače v označených transportních vzorkovnicích z plastu se vzduchotěsně uzavíratelným víkem
-
Programovatelný teplotní záznamník (logger)
-
Čistý nerezový plech nebo tác na manipulaci se vzorkovači
-
Plastové stahovací pásky
-
Ultračistá voda
-
Nylonové lano
-
Kotva nebo závaží na ukotvení vzorkovačů ke dnu
-
Bóje nebo plovák
-
Zařízení pro měření teploty, vodivosti a pH vody
-
Zařízení pro odběr vzorků vody - kbelík na laně nebo vzorkovnice na tyči
-
Vinylové rukavice
7.3.1.2 Postup rozmísťování vzorkovačů
1. Připravte si všechen materiál potřebný k instalaci vzorkovačů (7.3.1.1).
2. Otevřete vzorkovnici s terénní kontrolou (7.8.3), vyjměte kontrolní vzorkovač a položte jej na nerezový tác nebo na čisté víko transportní plastové nádoby. Zaznamenejte čas do protokolu o odběru (viz část 10; Obrázek 7).
3. Vyjměte vzorkovač ze vzorkovnice a pomocí stahovacích pásků jej upevněte na držák vzorkovacího zařízení (Obrázek 7).
4. K držáku vzorkovacího zařízení upevněte automatický záznamník teploty a aktivujte ho.
5. Po založení všech vzorkovačů do držáků vzorkovacího zařízení, vložte držáky se vzorkovači do ochranné klece z perforovaného plechu nebo do ochranné mříže, upevněte vzorkovací zařízení pomocí lana ke břehu (Obrázek 8) a vzorkovací zařízení ponořte co nejrychleji do vody.
6. Vzorkovací zařízení ukotvěte vhodně k říčnímu nebo jezernímu břehu nebo také ke dnu v místě odběru vzorků a ponoří se pod hladinu pomocí bóje nebo plováku.
7. Uzavřete všechny prázdné vzorkovnice a odložte je na čistém místě v laboratoři - budou opět použity při vyjmutí vzorkovačů po expozici.
8. Vložte terénní kontrolu (7.8.3) zpátky do její vzorkovnice. Zaznamenejte čas ukončení instalace vzorkovačů. Terénní kontrola je transportována zpátky do laboratoře a skladována při 4°C.

Obrázek 7. Instalace pasivních vzorkovačů v terénu. Plastová vzorkovnice na transport a uskladnění vzorkovače je na obrázku nahoře vlevo. Střední obrázek ukazuje umístění vzorkovačů do držáku, který se pak umístí do ochranné klece z perforovaného nerezového plechu (v pozadí). Na fotografii vpravo je demonstrováno umístění vzorkovačů v ochranných klecích do vody za pomocí lana.
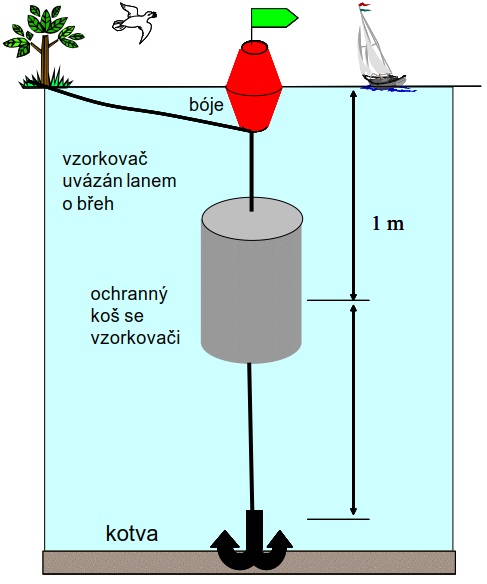
Obrázek 8. Příklad instalace vzorkovacího zařízení pasivních vzorkovačů v terénu. Vzorkovač může být v případě silného proudění vody ukotven na dno pomocí kotvy nebo závaží.

Obrázek 9. Pasivní vzorkovač před a po 28 denní expozici v povrchové vodě.
7.3.2 Vyjímání vzorkovačů
Při vyjímání vzorkovačů z vody po expozici platí všeobecná pravidla uvedená v normě ČSN EN ISO 5667-23 Jakost vod - Odběr vzorků - Část 23: Návod pro pasivní odběr vzorků v povrchových vodách [1].
7.3.2.1 Materiál potřebný při vyjímání pasivních vzorkovačů po expozici
-
Transportní vzorkovnice, v kterých byly vzorkovače transportovány na odběrové místo (viz obr. 7)
-
Čistý nerezový plech nebo tác na manipulaci se vzorkovači
-
Ultračistá voda
-
Skleněná kádinka
-
Zařízení pro měření teploty, vodivosti a pH vody
-
Odběrové zařízení na odběr vzorků vody - kbelík na laně nebo vzorkovnice na tyči
-
Vinylové rukavice
-
Papírové ubrousky
7.3.2.2 Postup vyjímání vzorkovačů po expozici
1. Otevřete vzorkovnici s terénní kontrolou (7.8.3) a pomocí pinzety vyjměte kontrolní vzorkovač a položte jej na čistý nerezový tác. Zaznamenejte čas do protokolu o odběru (viz část 10).
2. Vyjměte vzorkovací zařízení z vody. Zkontrolujte, zda vzorkovače během expozice nebyly poškozeny.
3. Dokumentujte situaci fotografií exponovaných vzorkovačů po jejich vyjmutí z vody (Obrázek 9).
4. Vyjměte exponovaný vzorkovač z držáku a položte jej do čistého nerezového plechu naplněného lokální vodou z místa odběru.
5. Použitím vody z odběrového místa a papírového ubrousku šetrně očistěte vzorkovače od vrstvy biofilmu za co nejkratší dobu.
6. Vložte terénní kontrolu (7.8.3) a exponované vzorkovače zpátky do jejich označených vzorkovnic.
7. Zaznamenejte čas ukončení instalace vzorkovačů.
8. Vzorkovače by měly být transportovány zpátky do laboratoře a skladovány při -20°C.
V závislosti na ročním období a místě expozice vzorkovače mohou být čisté nebo zarostlé vrstvou řas nebo jiných vodních organismů. Doporučuje se dokumentovat situaci fotografií exponovaných vzorkovačů po jejich vyjmutí z vody. Vzhledem k tomu, že difúzní vrstva hydrogelu není dále analyzována, není nutné důkladně čistit vzorkovač. Difúzní vrstva hydrogelu plní funkci ochrany hydrogelu s dispergovaným adsorbentem před znečištěním. Vzorkovače se vloží do stejné vzorkovnice, ve které byly přineseny na místo odběru a vzorkovnice se vzduchotěsně uzavřou. Vzorkovnice se umístí do chlazeného transportního kontejneru bez přístupu světla a transportují se co nejdřív do laboratoře.
|
|
Pozor! Čištění pasivních vzorkovačů je nejlepší provést bezprostředně po vyjmutí z vody, za použití vody z vzorkovaného vodního útvaru. Ponoření vzorkovačů do vody z místa odběru minimalizuje jejich expozici v ovzduší a související ztráty látek nebo kontaminaci vzorkovačů. Voda z odběrového místa obsahuje sledované látky v podobných koncentracích, jakým byl vzorkovač vystaven během expozice. Proto je lokální voda vhodnější na čištění, než například destilovaná voda z laboratoře. |

Obrázek 10. Ponorný záznamník teploty - připevní se pomocí stahovacího pásku na rám vzorkovacího zařízení při jeho expozici ve vodě.
7.4 Odeslání vzorků k analýze
Exponované vzorkovače by měly být zaslány do laboratoře ke zpracování v co nejkratším čase. Během přepravy se doporučuje udržovat vzorky při nízké teplotě (< 8°C) použitím tepelně izolovaných přepravních kontejnerů a chladících elementů. Je-li požadována kontrola teploty během přepravy, mohou být přidány do balíčku záznamníky teploty. Při odesílání vzorků by měla přijímající laboratoř být vždy předem informována. Na zásilce je potřeba uvést fyzickou adresu, ne P.O. Box. Je vhodné, spolu s adresou, uvést i kontaktní telefonní číslo. Odběrová zařízení, která nemusí být transportována expresně, mohou být odeslána odděleně pravidelnou poštovní službou, která je obvykle levnější.
7.5 Analytické stanovení perfluorovaných látek a vybraných léčiv v extraktech z pasivních vzorkovačů
7.5.1 Extrakce vzorkovačů
Všechny vzorky musí být během celého zpracování označeny a nesmí dojít k jejich záměně. Pokud byly vzorkovače uskladněny v chladničce, měly by být vyndány s dostatečným předstihem a temperovány na laboratorní teplotu. Po expozici jsou vzorkovače rozebrány čistými nástroji (šroubováky a klíče musí být čisté, odmaštěné a opláchnuté v hexanu, acetonu a methanolu) na čisté podložce z hliníkové fólie. Vrstvy sorbentového gelu jsou uloženy do skleněných vialek o objemu 25 ml (Obrázek 11). Mechanické narušení nebo zpřetrhání gelů na menší kusy nemá vliv na kvalitu extrakce. Zpracovány jsou pouze vrstvy gelu s dispergovaným hydrogelem, ostatní části se neanalyzují. Vrstvy gelu jsou následně zality methanolem o objemu alespoň 10 ml a v skleněné vialce uloženy na třepačku na dobu alespoň 24 hodin. V případě potřeby je možné přidat k vzorku roztok značených standardů některých látek pro kontrolu ztrát během zpracování vzorku. Je možné provést i opakovanou extrakci dalším alikvotním podílem methanolu, případně acetonu. Po extrakci je gel odstraněn z roztoku pinzetou a extrakt dále zpracováván. Pláty gelu zalité methanolem mohou být uskladněny v mrazničce při -20°C. Maximální doba skladování závisí na vlastnostech cílových analytů a je vhodné provést test stability sledovaných látek v methanolu. Vhodnější je však skladovat čistý extrakt. Perfluorované látky jsou perzistentní a zpravidla nepodléhají při skladování rozkladu. Pro léčiva je typická vysoká variabilita od relativně nestabilních sloučenin, jako jsou antibiotika až po sloučeniny vysoce stabilní, jako je např. karbamazepin.
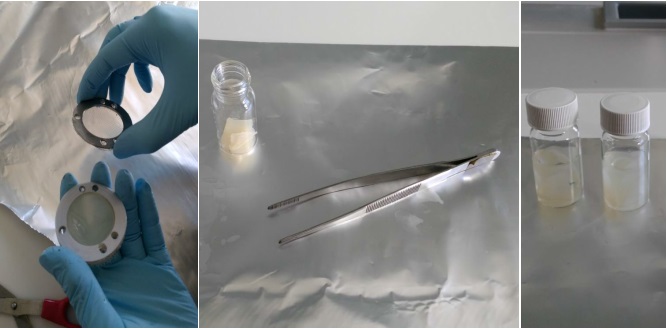
Obrázek 11. Extrakce analytů z hydrogelových vrstev vzorkovače s dispergovaným adsorbentem.
Objem extraktu je dále nutné zredukovat pod proudem dusíku při maximální teplotě 40°C do poslední kapky. Poté je přidáno požadované rozpouštědlo vhodné pro následnou instrumentální analýzu (v tomto případě methanol) do konečného objemu ideálně 1 ml. Sledované sloučeniny nejsou těkavé, proto nehrozí ztráty jejich odpařováním z roztoku při redukci objemu rozpouštědla. Jelikož se jedná o polární a netěkavé látky, není nutné používat značený standard pro kontrolu úniku látky odparem. U některých zcela fluorovaných látek, zejména s délkou řetězce nad 10 atomů uhlíku, je větším problémem potenciální sorpce látek na stěny nádob. Před vlastním analytickým stanovaním se odebere alikvot vzorku, naředí se do rozpouštědla odpovídajícího počátečnímu složení mobilní fáze při analytickém stanovení (v případě perfluorovaných látek do směsi methanolu s voudou 1:1) a přidá se (ručně, nebo automaticky) roztok izotopicky značeného instrumentálního vnitřního standardu.
7.5.2 Instrumentální analýza
Pro vlastní analýzu je možné použít postupů, které jsou v laboratoři zavedené a validované nebo odeslat extrakt vzorku k analýze do jiné laboratoře. Perfluorované organické látky a vybraná farmaka je možné v extraktech vzorkovačů stanovit bez dalších čistících kroků pomocí kapalinové chromatografie s tandemovou hmotnostní detekcí (LC/MS). Pro zjištění analytické výtěžnosti (kontrola ztrát analytu během přípravy vzorku) se využívá izotopicky značených standardů, jejichž známá hmotnost se přidá do vzorků před počátkem přípravy vzorku. Výsledky jsou uváděny s příslušnou výtěžností. Obecně jsou meze stanovení jednotlivých analytů závislé spíše na ionizačních vlastnostech dané sloučeniny a na obsahu interferující matrice ve vzorku než na výsledném objemu vzorku. Naměřené výsledky představují koncentrace polárních perfluorovaných látek v extraktu vzorku, výsledky je ještě nutno přepočítat na množství analytu ve vzorku dle postupu přípravy vzorku.
7.6 Kalibrační parametry vzorkovače
Za účelem přepočítání množství akumulované chemické látky ve vzorkovači na koncentraci ve vodě je potřebné znát parametry pasivního vzorkovače pro sledované látky: a) difúzní koeficienty látek v agarozovém hydrogelu, aby bylo zřejmé, zda přestup látek do vzorkovače je kontrolován jejich difúzí v hydrogelu, nebo difúzí v mezní vrstvě vody na povrchu vzorkovače; b) distribuční koeficienty látek v systému adsorbent-voda pro případ, že během expozice dojde v systému vzorkovač-voda k ustálení distribuční rovnováhy.
Hodnota distribučního koeficientu látky v systému vzorkovač-voda (KSW) kvantifikuje poměr koncentrace sledované látky v adsorbentu vzorkovače a její volně rozpuštěné koncentrace ve vodné fázi v rovnovážném stavu. Hodnoty KSW jsou potřebné pro přepočet koncentrace látek v pasivním vzorkovači na koncentraci ve vodné fáze. Stanovení se provádí v laboratorních podmínkách měřením koncentračního poměru látky v adsorbentu, a v zvoleném objemu vody, do které byly přidány sledované látky, po ustálení distribuční rovnováhy sorbentu s vodní fází.
|
|
Pozor! Součástí metodiky jsou hodnoty distribučních koeficientů perfluorovaných látek a vybraných farmak mezi různými adsorbenty a vodou (kapitola 11), které byly stanoveny jako součást řešení projektu „Emergentní polutanty ve složkách životního prostředí“ (TB030MZP001) podpořeného TA ČR v rámci programu BETA. |
|
Součástí metodiky jsou i hodnoty difúzních koeficientů perfluorovaných látek a vybraných farmak v agarozovém hydrogelu (kapitola 12), které byly stanoveny jako součást řešení projektu „Emergentní polutanty ve složkách životního prostředí“ (TB030MZP001) podpořeného TA ČR v rámci programu BETA. |
7.7 Interpretace výsledků měření
Výpočet koncentrace sledované látky ve vodě z množství ve vzorkovači zahrnuje několik kroků, které jsou podrobně popsány níže. Pro odhad koncentrace sledované látky z jejího množství v pasivním vzorkovači je potřeba znát vzorkovací rychlost RS (specifický parametr pro každou látku, který vyjadřuje ekvivalentní objem vody, navzorkovaný za jednotku času) a distribuční koeficient látky v systému sorbent-voda Ksw [41]. Ve zkratce se uplatňuje tento postup: Z množství látky akumulované ve vzorkovači, doby expozice, vzorkovací rychlosti RS a distribučního koeficientu látky v systému vzorkovač-voda KSW, se vypočítá pomocí ověřeného modelu koncentrace látky ve vodě.
Koncentrace ve vodě se dají vypočítat z množství látky sorbovaného ve vzorkovači (NS), ze vzorkovací rychlosti pro danou látku RS a z distribučního koeficientu v systému sorbent-voda KSW:

cW je koncentrace ve vodě (ng l-1)
NS je množství látky akumulované ve vzorkovači (ng)
N0 je množství látky v laboratorní kontrole (7.8.2) (ng)
mS je hmotnost sorbentu ve vzorkovači (kg)
KSW je distribuční koeficient sorbent/voda (l kg-1)
t je doba expozice (d)
Pro kinetické vzorkovače, které pracují v lineárním akumulačním módu je člen v hranatých závorkách přibližně rovný (Rs×t)/(Ksw×ms), a koncentraci ve vodě lze vypočítat jako:

Jmenovatelé v rovnicích 2 a 3 se dají interpretovat jako zdánlivý objem vody, ze kterého vzorkovač odstraní analyt po dobu expozice (Obrázek 2). V případě rovnovážného vzorkování je tento objem omezen sorpční kapacitou vzorkovače (Ksw×ms). Při kinetickém vzorkování je zdánlivý exktrahovaný objem vody omezen vzorkovací rychlostí a expozičním časem (RS×t).
7.7.1 Příklad stanovení rozpuštěné koncentrace perfluorovaných látek v povrchové vodě pomocí pasivního vzorkování
Následující výpočet je příkladem výpočtu koncentrace ve vodě z množství látky zachycené v pasivním vzorkovači během terénní aplikace. Pasivní vzorkovač připravaný podle postupu uvedeného v kapitole 0 byl exponován v povrchové vodě na lokalitě Bělov v řece Morava od 4. 4. 2016 do 2. 5. 2016. Vzorkovač se skládá ze dvou prstenů z nerezové oceli, mezi nimiž jsou umístěny čtyři vrstvy gelu o tloušťce 0,1 cm. Vnější (difuzní) vrstvy tvoří 1,5% agarózový hydrogelový film o průměru 5,6 cm s vnořenou nylonovou sítí. Vnitřní (sorpční) vrstvy jsou tvořeny 1,5% agarózovém hydrogelem o průměru 3,8 cm s rozptýleným sorbentem Oasis HLB®. Aktivní plocha vzorkovače je přibližně 22,68 cm2 a hmotnost sorbentu ve vzorkovači je 0,1134 g. Doba expozice byla 28 dní. Teplota vody kolísala mezi 8,0°C a 13,5°C s průměrnou hodnotou 11°C.
Vzorkovací rychlost byla získána laboratorní kalibrací (viz kapitola 13) s použitím vzorkovače o aktivní ploše 3,14 cm2. Vynesením závislosti sorbovaného množství na jednotku hmotnosti sorbentu v čase Vynesením závislosti a proložením bodů přímkou byla získána rovnice lineárního vzorkování (Obrázek 12). Ze směrnice regresní přímky, množství sorbentu v kalibračním vzorkovači (0,0245 g) a průměrné koncentrace ve vodě během kalibrace (PFOA: 0,270 ng ml-1; PFOS: 0,392 ng ml-1) byly vypočítány rychlosti vzorkování RS a následně normalizovány na povrch vzorkovače použitého v terénu (Rovnice 4).

RS je rychlost vzorkování pro sledovanou látku (ml h-1)
β0 je směrnice kalibrační závislosti (ng g-1 d-1)
mS je hmotnost sorbentu (0,0245 g)
cW je koncentrace ve vodě (ng ml-1)
At je plocha vzorkovače použitého v terénu (cm2)
A0 je plocha vzorkovače použitého při kalibraci (cm2)
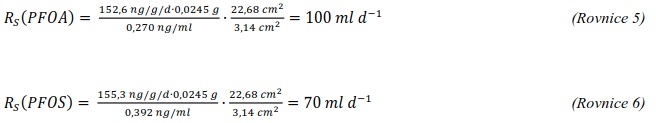
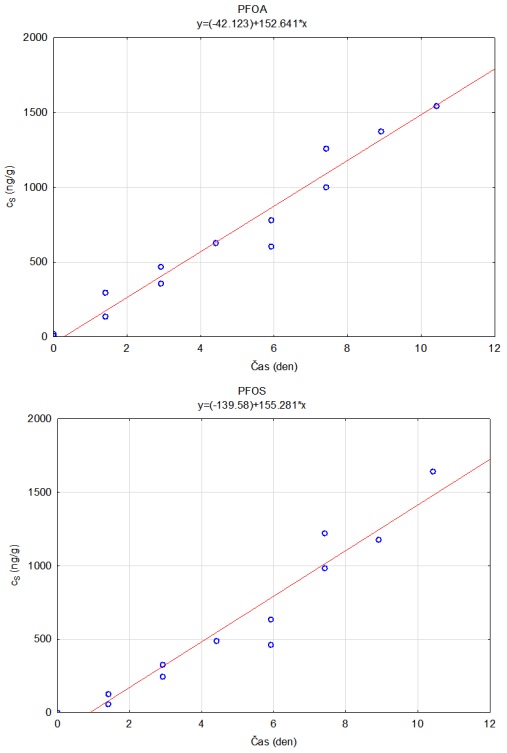
Obrázek 12. Příklady laboratorních kalibračních křivek pasivního vzorkovače, zobrazující lineární nárůst koncentrace látky PFOA a PFOS v čase v pasivním vzorkovači s difúzní vrstvou hydrogelu exponovaném v roztoku s konstantní koncentrací látky ve vodě.
Stejným způsobem je možné vypočítat rychlost vzorkování pro každou látku použitou v kalibraci. Se známými vzorkovacími rychlostmi, dobou expozice v terénu a množstvím látky ve vzorkovači je následně možné vypočítat časově vážený průměr koncentrace ve vodě po dobu expozice (Rovnice 2). V případě dlouhých expozic se může u některých látek ustálit distribuční rovnováha mezi sorbentem a vodou, a tedy akumulace látky může být nelineární. Proto je pro výpočet nutné znát i distribuční koeficient mezi použitým sorbentem a vodou (KSW).
Pokud je ve vzorku terénní kontroly naměřené množství sledovaných látek menší než 10% hodnoty v exponovaných vzorkovačích, je možné při výpočtu tuto hodnotu odečíst od množství látky ve vzorkovači. Při hodnotách nad 10% je nutné zvážit vhodnost použité metody. V případě PFOA a PFOS byly hodnoty na úrovni, respektive pod úrovní meze stanovení.

cW je koncentrace ve vodě (ng l-1)
NS je množství látky vo vzorkovači (pg)
mS je hmotnost sorbentu ve vzorkovači (0,1134 g)
KSW je distribuční koeficient sorbent/voda (ml g-1)
t je doba expozice (28 d)

Vypočtené koncentrace vyjadřují časově vážený průměr (cTWA) a v tomto případě souhlasí s koncentracemi zjištěnými v bodových odběrech vzorků vody na začátku (c1) a na konci (c2) expozice (Tabulka 3)1.
Tabulka 3. Porovnání koncentrací zjištěných pasivním vzorkováním (CTWA) a bodovými odběry na lokalitě Bělov v řece Morava před a po ukončení pasivního vzorkování (C1 a C2). Hodnoty jsou v ng l-1.
|
Látka |
CTWA |
C1 |
C2 |
|
PFOA |
0,52 |
0,42 |
0,75 |
|
PFOS |
1,83 |
1,24 |
2,03 |
7.8 Zajištění a řízení kvality (QA/QC)
Pro zajištění kvality monitorování pomocí pasivního vzorkování je zapotřebí řada opatření řízení kvality. Patří mezi ně analýza rozpouštědel a použitých činidel (činidel pro slepé stanovení), kontroly, terénní kontroly a zařízení pro určení výtěžnosti. Porovnání činidel pro slepé stanovení, laboratorních a terénních kontrol může pomoci identifikovat možné zdroje kontaminace, a podniknout potřebná opatření k zajištění kvality.
7.8.1 Činidlo pro slepé stanovení (reagent blank)
Podle normy ČSN EN ISO 5667-23 [1] se takto označuje alikvotní objem činidla, používaného při úpravě pasivních vzorkovacích zařízení, který se po nasazení vzorkovacího zařízení analyzuje za účelem jakéhokoli znečištění použitými činidly. Množství látky, nalezeno v tomto typu slepého vzorku je možno odečíst od množství nalezeného v exponovaných vzorkovačích, laboratorních a terénních kontrolách.
7.8.2 Laboratorní kontrola
Pojem laboratorní kontroly není přímo definován v normě ČSN EN ISO 5667-23 [1]. Laboratorní kontrola označuje pasivní vzorkovací zařízení pro řízení kvality, určené pro zaznamenávání jakékoliv chemické látky akumulované v pasivních vzorkovacích zařízeních během výroby, sestavování (montáže), skladování a následné analýzy. Pro vzájemnou porovnatelnost dat mezi exponovaným vzorkovačem a laboratorní kontrolou je nutné, aby laboratorní kontrolní vzorky byly připraveny v jedné homogenní várce spolu se vzorkovači určenými pro terénní nasazení. Množství analyzovaných látek v laboratorních kontrolních vzorcích by v ideálním případě mělo být porovnatelné s množstvím nalezeným v činidle pro slepé stanovení. Výsledky stanovení sledovaných látek v laboratorních kontrolách lze použít pro odhad meze detekce (LOD) nebo meze stanovení (LOQ) metody. Za tímto účelem se množství analytu nalezené v laboratorní kontrole 7.8.2 nebo v instrumentálním slepém vzorku dosadí do rovnice 2. Pokud je koncentrace sledované sloučeniny v laboratorní kontrole mnohem vyšší než v činidle pro slepé stanovení (v slepém vzorku), postup přípravy vzorkovače by měl být kriticky zhodnocen a měla by být identifikována a odstraněna příčina těchto zvýšených koncentrací [17].
|
|
Pozor! Vzhledem k tomu, že hodnoty LOQ pro koncentrace vody jsou vypočteny s použitím vzorkovací rychlosti, která je specifická pro každý odběr, hodnoty LOQ jsou taky specificky vázané na konkrétní odběr. Integrační charakter pasivního vzorkování často umožňuje snížit LOQ prodloužením expoziční doby vzorkovače. |
7.8.3 Terénní kontrola
Podle normy ČSN EN ISO 5667-23 [1] označuje pojem terénní kontrola pasivní vzorkovací zařízení pro řízení kvality, určené pro zaznamenávání jakékoliv chemické látky akumulované v pasivních vzorkovacích zařízeních během výroby, sestavování (montáže), skladování, dopravy, rozmístění, vyjmutí a následné analýzy. Terénní kontrolní vzorky mohou obsahovat vyšší koncentrace sledované látky než laboratorní kontrolní vzorky, zvláště pokud jsou vzorkovače osazovány ve vysoce kontaminovaných oblastech v blízkosti továren, dálnic, na palubách lodí, nebo když je instalace vzorkovačů prováděna v oblasti se zvýšeným znečištěním ovzduší. Vzhledem k tomu, že kontaminace terénních kontrolních vzorků je specifická pro konkrétní odběr, nedoporučuje se obecně používat tyto vzorky řízení kvality pro stanovení průměrné hodnoty slepých stanovení nebo mezí stanovení. Terénní kontroly by mělo být především použity ke kvalitativnímu posouzení kontaminace z ovzduší během přepravy a procesu nasazení/vyjmutí vzorkovačů v terénu. Zvýšené koncentrace analytů v terénních kontrolách může naznačit potřebu revidovat/změnit postup instalace a vyjmutí pasivních vzorkovačů v terénu.
7.8.4 Zařízení pro určení výtěžnosti (recovery spike)
Podle normy ČSN EN ISO 5667-23 [1] se takto označuje pasivní vzorkovací zařízení, k němuž byla přidána známá hmotnost standardu pro určení analytické výtěžnosti, které se používá ke stanovení výtěžnosti znečišťující látky z pasivních vzorkovacích zařízení po expozici.
7.8.5 Použití kontrolních a slepých vzorků
Množství sledovaných látek, naměřených v činidle pro slepé stanovení (7.8.1) může být odečteno od množství změřeného v exponovaných vzorcích, laboratorních (7.8.2) a terénních kontrolních vzorcích (7.8.3). Množství sledované látky v laboratorní kontrole by mělo být nejlépe podobné množství změřenému v činidle pro slepé stanovení. Výsledky měření laboratorních kontrolních vzorků se mohou použít k odhadu meze detekce a meze stanovení metody (LOD a LOQ). Pokud je množství sledované látky v laboratorní kontrole mnohem vyšší než v činidle pro slepé stanovení, postup přípravy vzorkovače by měl být kriticky posouzen a měly by být identifikovány a odstraněny příčiny těchto zvýšených koncentrací (a následné vyšší LOD/LOQ). Korekce množství sledované látky v exponovaném vzorkovači (Nt) použitím laboratorní kontroly (N0) je založeno na odčítání. Orientačním pravidlem zůstává, že je akceptovatelné, když je množství látky v laboratorní nebo terénní kontrole menší než 10% množství přítomného v exponovaném vzorkovači. V takovém případě není nutná ani korekce množství látky v exponovaném vzorkovači.
7.8.6 Řízení kvality v přípravě vzorků na toxikologickou analýzu
Kromě chemické analýzy lze pasivních vzorkovačů využít i k sledování toxických účinků směsí emergentních látek, které se akumulují ve vzorkovači během expozice. Toxicita extraků vzorkovačů se sleduje různými biotesty. Řízení kvality pro pasivní vzorkovače určených pro toxikologické analýzy je podobné těm pro chemickou analýzu. Laboratorní kontrolní vzorky (7.8.2) a terénní kontrolní vzorky (7.8.3) jsou zpracovány společně s exponovanými vzorkovači a zachází se s nimi stejně jako s ostatními vzorky. Slepé vzorky (7.8.1) se zpracují stejně jako ostatní vzorky, ale vzorek neobsahuje adsorbent vzorkovače. Zatímco výtěžnost toxicity je dobrý způsob, jak testovat pro obecnou použitelnost metodou extrakce a dalšího čištění a zpracování vzorků, v případě nedostatečné výtěžnosti očekávaného toxického efektu v zpracovaném vzorku je problematické dopátrat se příčiny [39]. Proto se doporučuje, aby kromě testu výtěžnosti toxicity, byly analyzovány i vzorky pro stanovení výtěžnosti (7.8.4). Sada sloučenin, používaných k monitorování výtěžnosti metody by měla obsahovat takové chemické látky, o kterých je známo, že způsobují toxický efekt, sledovaný v extraktech vzorkovačů zvoleným toxikologickým testem. Tento přístup pomůže identifikovat skupiny látek a jejich vlastnosti, které by mohly být zodpovědné za nedostatečnou účinnost extrakce látek způsobujících sledovaný toxický účinek [42].
7.9 Zpráva o zkoušce
Zpráva o získaných odhadech koncentrace ve vodné fázi by měla obsahovat informace o místě expozice, způsobu odběru vzorků a údaje o použitých metodách analýzy vzorků a souvisejících vzorcích řízení kvality, jakož i podrobnosti o výpočtech, které byly použity. Tato informace je důležitá, aby umožnila uživatelům data přepočítat v případě, že by budoucnosti byly zavedeny nové metody výpočtu koncentrací ve vodní fázi, nebo byly dostupné kvalitnější kalibrační parametry [40]. Norma ČSN ISO 56667-23 [1] specifikuje minimální rozsah informací, které by měly být uvedeny ve zprávě.
8 Popis novosti a uplatnění metodiky
Metodika je inovativní v několika aspektech. Novost představuje konstrukce pasivního vzorkovače, ve kterém je transport látky do vzorkovače kontrolován difúzí ve vrstvě v hydrogelu, což umožňuje stanovení perfluorovaných látek a vybraných farmak ve vodním prostředí s definovanou nejistotou měření. Pro vyvinutý vzorkovač jsou v metodice uvedeny i kalibrační parametry (difúzní koeficienty perfluorovaných látek a tří farmak v difúzním hydrogelu a distribuční koeficienty perfluorovaných látek a tří farmak v systému adsorbent-vodní roztok pro různé adsorbenty), které umožňují odhad koncentrací látek ve vodní fázi z množství akumulovaného ve vzorkovači.
Tato metodika má pomoci uživatelům pasivního vzorkování, kteří chtějí realizovat pasivní odběr perfluorovaných látek a dalších polárních organických látek ve svém výzkumu nebo při monitorování znečišťujících látek vodního prostředí. Je určena pro uživatele pasivních vzorkovačů, zejména pro firmy a instituce zabývající se kvalitou a znečištěním vodního ekosystému. V podmínkách ČR jsou to zejména státní podniky Povodí, Český hydrometeorologický ústav a společnosti zodpovědné za kvalitu pitné vody.
Metodika je určena i pro orgány státní správy, zejména Ministerstva životního prostředí. Metodika přispívá k plnění Koncepce monitoringu perzistentních organických polutantů (POPs) a zpracování a využití dat o POPs v ČR a rovněž k implementaci Stockholmské úmluvy o perzistentních organických polutantech, zejména k hodnocení účinnosti přijatých opatření vedoucích ke snížení výskytu vybraných perfluorovaných látek ve vodním prostředí. Pro sledování efektivity opatření Stockholmské úmluvy v delším časovém horizontu byl ustaven další instrument, a to Globální monitorovací plán (GMP) SÚ, který určuje, které matrice je třeba prioritně sledovat. Mezi tyto klíčové matrice patří pro všechny zahrnuté látky volné ovzduší, mateřské mléko a krev, pro více rozpustné látky (zejména PFOS) pak také voda. U vody jako nové matrice GMP je třeba ověřit, které ze současně používaných odběrových technik jsou nejvhodnější pro sledování perfluorovaných látek [43].
Zároveň má metodika přispět i k implementaci Vodní rámcové směrnice 2000/60/ES [44] při hodnocení chemického stavu vodních útvarů podle Evropské směrnice 2013/39/EU [45]. Perfluoroktansulfonová kyselina a její deriváty (PFOS) jsou aktuálně zařazeny na seznam prioritních nebezpečných látek ve vodním prostředí. Metodika může být vhodně využita při hodnocení chemického stavu vodních útvarů podle Evropské směrnice 2013/39/EU, k analýze dlouhodobých trendů v monitorovaných vodních útvarech s cílem hodnocení účinnosti přijatých opatření vedoucích ke snížení výskytu perfluorovaných látek ve vodním prostředí. Směrnice 2013/39/EU zmiňuje potřebu vývoje nových nástrojů skýtajících potenciál pro budoucí využití, jako je pasivní odběr vzorků, pro monitorování prioritních a specifických znečišťujících látek ve vodním prostředí. Pasivní vzorkovače lze využít jako doplňkové metody jak pro situační, tak i pro provozní a průzkumné monitoringy [46,47].
Metodika se kromě perfluorovaných látek zaměřuje i na vzorkování vybraných farmak (diklofenak, karbamazepin a sulfomethoxazol). V roce 2015 byl diklofenak prováděcím rozhodnutím Evropské komise 2015/495 zařazen na seznam sledovaných látek pro monitorování v rámci celé Unie v oblasti vodní politiky podle směrnice Evropského parlamentu a Rady 2008/105/ES.
Evropská asociace laboratoří NORMAN (www.norman-network.net) v spolupráci s komunitou vědců zabývajících se problematikou znečištění vod chemickými látkami SOLUTIONS (www.solutionsproject.eu) formulovala stanovisko k plánované aktualizaci Vodní rámcové směrnice v roce 2019 [48]. Jedním z hlavních doporučení v stanovisku je návrh na doplnění stávajícího monitoringu bioakumulativních látek pasivním vzorkováním za účelem snížení nejistot vzorkování a jednoznačnější klasifikace stavu vodních útvarů. K naplnění tohto cíle má sloužit i tato metodika.
9 Literatura
[1] ČSN EN ISO 5667-23 Jakost vod - Odběr vzorků - Část 23: Návod pro pasivní odběr vzorků v povrchových vodách, Úřad pro Tech. Norm. Metrol. a Státní Zkušebnictví. (2011). http://www.tzbinfo.cz/normy/csn-en-iso-5667-23-2011-09.
[2] D.A. Alvarez, J.D. Petty, J.N. Huckins, T.L. Jones-Lepp, D.T. Getting, J.P. Goddard, et al., Development of a Passive, in Situ, Integrative Sampler for Hydrophilic Organic Contaminants in Aquatic Environments, Environ. Toxicol. Chem. 23 (2004) 1640-1648.
[3] C. Harman, I.J. Allan, E.L.M. Vermeirssen, Calibration and use of the polar organic chemical integrative sampler--a critical review., Environ. Toxicol. Chem. 31 (2012) 2724-38. doi:10.1002/etc.2011.
[4] G. Poulier, S. Lissalde, A. Charriau, R. Buzier, F. Delmas, K. Gery, et al., Can POCIS be used in Water Framework Directive (2000/60/EC) monitoring networks? A study focusing on pesticides in a French agricultural watershed, Sci. Total Environ. 497-498 (2014) 282-292. doi:10.1016/j.scitotenv.2014.08.001.
[5] P. Mayer, J. Tolls, L. Hermens, D. Mackay, Equilibrium Sampling Devices, Environ. Sci. Technol. 37 (2003) 184A-191A.
[6] EU, Directive 2000/60/EC of the European parliament and of the council of 23 October 2000 establishing a framework for community action in the field of water policy., Off. J. Eur. Union. L327 (2000) 1-72.
[7] EU, Directive 2008/105/EC of the the European parliament and of the council of 16 December 2008 on environmental quality standards in the field of water policy., Off. J. Eur. Union. L348 (2008) 84-96. http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2008:348:0084:0097:EN:PDF.
[8] EU, Directive 2013/39/EU of the European Parlament and of the Council of 12 August 2013 amending Directives 2000/60/EC and 2008/105/EC as regards priority substances in the field of water policy, Off. J. Eur. Union. L226 (2013) 1-17.
[9] B. Vrana, I.J. Allan, R. Greenwood, G.A. Mills, E. Dominiak, K. Svensson, et al., Passive sampling techniques for monitoring pollutants in water, TrAC Trends Anal. Chem. 24 (2005) 845- 868.
[10] F. Stuer-Lauridsen, Review of passive accumulation devices for monitoring organic micropollutants in the aquatic environment, Environ. Pollut. 136 (2005) 503-524. doi:10.1016/j.envpol.2004.12.004.
[11] G. Ouyang, J. Pawliszyn, Configurations and calibration methods for passive sampling techniques, J. Chromatogr. A. 1168 (2007) 226-235. doi:10.1016/j.chroma.2007.01.133.
[12] A. Kot-Wasik, B. Zabiegała, M. Urbanowicz, E. Dominiak, A. Wasik, J. Namieśnik, Advances in passive sampling in environmental studies, Anal. Chim. Acta. 602 (2007) 141-163. doi:10.1016/j.aca.2007.09.013.
[13] J. Namieśnik, B. Zabiegała, A. Kot-Wasik, M. Partyka, A. Wasik, Passive sampling and/or extraction techniques in environmental analysis: a review., Anal. Bioanal. Chem. 381 (2005) 279- 301. doi:10.1007/s00216-004-2830-8.
[14] F.A. Esteve-Turrillas, A. Pastor, V. Yusà, M. de la Guardia, Using semi-permeable membrane devices as passive samplers, TrAC - Trends Anal. Chem. 26 (2007) 703-712. doi:10.1016/j.trac.2007.05.006.
[15] T. Lobpreis, B. Vrana, K. Dercová, Innovative approach to monitoring organic contaminants in aqueous environment using passive sampling devices, Chem. List. 103 (2009) 548-558.
[16] M.J. Lydy, P.F. Landrum, A.M. Oen, M. Allinson, F. Smedes, A.D. Harwood, et al., Passive sampling methods for contaminated sediments: state of the science for organic contaminants., Integr. Environ. Assess. Manag. 10 (2014) 167-78. doi:10.1002/ieam.1503.
[17] R. Lohmann, K. Booij, F. Smedes, B. Vrana, Use of passive sampling devices for monitoring and compliance checking of POP concentrations in water, Environ. Sci. Pollut. Res. 19 (2012) 1885- 1895.
[18] B. Vrana, E.L.M. Vermeirssen, I. Allan, J. Kohoutek, K. Kennedy, G. Mills, et al., Passive sampling of emerging compounds in the environment: state of the art and perspectives, (2010). http://www.norman-network.net/sites/default/files/files/Events/2009/2009May27-PraguePassiveSampling/norman_position_paper_pas_sampling.pdf.
[19] G.A. Mills, B. Vrana, I. Allan, D.A. Alvarez, J.N. Huckins, R. Greenwood, Trends in Monitoring Pharmaceuticals and Personal-Care Products in the Aquatic Environment by Use of Passive Sampling Devices, Anal. Bioanal. Chem. 387 (2007) 1153-1157.
[20] H. Söderström, R.H. Lindberg, J. Fick, Strategies for monitoring the emerging polar organic contaminants in water with emphasis on integrative passive sampling, J. Chromatogr. A. 1216 (2009) 623-630. doi:10.1016/j.chroma.2008.08.030.
[21] K. Booij, Performance of passive samplers for monitoring priority substances., (2009). http://www.ices.dk/reports/MHC/2009/MCWG09.pdf.
[22] K. Booij, C.D. Robinson, R.M. Burgess, P. Mayer, C.A. Roberts, L. Ahrens, et al., Passive sampling in regulatory chemical monitoring of nonpolar organic compounds in the aquatic environment., Environ. Sci. Technol. 50 (2016) 3-17. doi:10.1021/acs.est.5b04050.
[23] OSPAR Joint Assessment and Monitoring Programme (JAMP) 2014−2021. Update 2015 (Agreement 2014−02) , London, 2015., (2015). http://www.ospar.org/documents?d=32988.
[24] C. Miège, N. Mazzella, I. Allan, V. Dulio, F. Smedes, C. Tixier, et al., Position paper on passive sampling techniques for the monitoring of contaminants in the aquatic environment - Achievements to date and perspectives, Trends Environ. Anal. Chem. 8 (2015) 20-26. doi:10.1016/j.teac.2015.07.001.
[25] R. Greenwood, G.A. Mills, B. Vrana, I. Allan, R. Aguilar-Martínez, G. Morrison, Monitoring of priority pollutants in water using Chemcatcher passive sampling devices, in: R. Greenwood, G. Mills, B. Vrana (Eds.), Compr. Anal. Chem. Vol. 48, Elsevier, Amsterdam, 2007: pp. 199-229.
[26] K. Booij, B. Vrana, J.N. Huckins, Theory, modelling and calibration of passive samplers used in water monitoring, in: R. Greenwood, G. Mills, B. Vrana (Eds.), Compr. Anal. Chem. 48. Passiv.
Sampl. Tech. Environ. Monit., Elsevier, Amsterdam, 2007: pp. 141-169. doi:10.1016/S0166526X(06)48007-7.
[27] J.N. Huckins, J.D. Petty, J.A. Lebo, F. V Almeida, K. Booij, D.A. Alvarez, et al., Development of the permeability/performance reference compound (PRC) approach for in situ calibration of semipermeable membrane devices(SPMDs)., Environ. Sci. Technol. 36 (2002) 85-91.
[28] H.H. Liu, C.S. Wong, E.Y. Zeng, Recognizing the limitations of performance reference compound (PRC)-calibration technique in passive water sampling, Environ. Sci. Technol. 47 (2013) 10104- 10105. doi:10.1021/es403353d.
[29] A. Vallejo, A. Prieto, M. Moeder, A. Usobiaga, O. Zuloaga, N. Etxebarria, et al., Calibration and field test of the Polar Organic Chemical Integrative Samplers for the determination of 15 endocrine disrupting compounds in wastewater and river water with special focus on performance reference compounds (PRC), Water Res. 47 (2013) 2851-2862. doi:10.1016/j.watres.2013.02.049.
[30] S.L. Kaserzon, D.W. Hawker, K. Booij, D.S. O’Brien, K. Kennedy, E.L.M. Vermeirssen, et al., Passive sampling of perfluorinated chemicals in water: in-situ calibration., Environ. Pollut. 186 (2014) 98-103. doi:10.1016/j.envpol.2013.11.030.
[31] R. Greenwood, G.A. Mills, B. Vrana, Passive sampling techniques in environmental monitoring, Elsevier, Amsterdam, 2006.
[32] A. Charriau, S. Lissalde, G. Poulier, N. Mazzella, R. Buzier, G. Guibaud, Overview of the Chemcatcher® for the passive sampling of various pollutants in aquatic environments Part A: Principles, calibration, preparation and analysis of the sampler, Talanta. 148 (2016) 556-571. doi:10.1016/j.talanta.2015.06.064.
[33] N. Morin, C. Miège, M. Coquery, J. Randon, Chemical calibration, performance, validation and applications of the polar organic chemical integrative sampler (POCIS) in aquatic environments, TrAC - Trends Anal. Chem. 36 (2012) 144-175. doi:10.1016/j.trac.2012.01.007.
[34] A. Charriau, S. Lissalde, G. Poulier, N. Mazzella, R. Buzier, G. Guibaud, Overview of the Chemcatcher® for the passive sampling of various pollutants in aquatic environments Part A: Principles, calibration, preparation and analysis of the sampler, Talanta. 148 (2016) 556-571. doi:10.1016/j.talanta.2015.06.064.
[35] ISO, Water quality - sampling - part 23: Guidance on passive sampling in surface waters ISO 5667-23:2011, (2011).
[36] Guidelines for the Use of the Semipermeable Membrane Device (SPMD) and the Polar Organic Chemical Integrative Sampler (POCIS) in Environmental Monitoring Studies, (n.d.). . http://pubs.usgs.gov/tm/tm1d4/pdf/tm1d4.pdf.
[37] POCIS, (n.d.). https://en.wikipedia.org/wiki/Polar_organic_chemical_integrative_sampler.
[38] R. Grabic, K. Grabicová, G. Fedorova, O. Golovko, T. Randák, Metodika sledování kontaminace povrchových vod organickými cizorodými látkami pomocí pasivních vzorkovačů. Edice metodik ; 158. Technologická řada, Jihočeská univerzita v Českých Budějovicích, Fakulta rybářství a ochrany vod, Vodňany, 2015.
[39] R.M. Burgess, Guidelines for Using Passive Samplers to Monitor Organic Contaminants at Superfund Sediment Sites. EPA/600/R-11/115, Washington, DC, 2012. https://cfpub.epa.gov/si/si_public_record_report.cfm?dirEntryId=238596 (accessed August 21, 2016).
[40] F. Smedes, K. Booij, Guidelines for passive sampling of hydrophobic contaminants in water using silicone rubber samplers, International Council for the Exploration of the Sea, Copenhagen, 2012. http://info.ices.dk/pubs/times/times52/120621 TIMES 52 Final.pdf.
[41] K. Booij, H.M. Sleiderink, F. Smedes, Calibrating the Uptake Kinetics of Semipermeable Membrane Devices using Exposure Standards., Environ. Toxicol. Chem. 17 (1998) 1236-1245.
[42] W. Brack, S. Ait-Aissa, R.M. Burgess, W. Busch, N. Creusot, C. Di Paolo, et al., Effect-directed analysis supporting monitoring of aquatic environments — An in-depth overview, Sci. Total Environ. 544 (2016) 1073-1118. doi:10.1016/j.scitotenv.2015.11.102.
[43] J. Weiss, J. de Boer, U. Berger, D. Muir, T. Ruan, A. Torre, et al., PFAS analysis in water for the Global Monitoring Plan of the Stockholm Convention. Set-up and guidelines for monitoring, United Nations Environment Programme (UNEP), Geneva, 2015.
[44] Evropská_unie, SMĚRNICE EVROPSKÉHO PARLAMENTU A RADY 2000/60/ES ze dne 23.
října 2000, kterou se stanoví rámec pro činnost Společenství v oblasti vodní politiky, Úřední Věstník Evr. Unie. L 327 (2000) 275-346.
[45] Evropská_unie, SMĚRNICE EVROPSKÉHO PARLAMENTU A RADY 2013/39/EU ze dne 12. srpna 2013, kterou se mění směrnice 2000/60/ES a 2008/105/ES, pokud jde o prioritní látky v oblasti vodní politiky, Úřední Věstník Evr. Unie. L226 (2013) 1-17.
[46] G. Hanke, P. Lepom, P. Quevaviller, eds., COMMON IMPLEMENTATION STRATEGY FOR THE WATER FRAMEWORK DIRECTIVE (2000/60/EC) Technical Report - 2009 - 025 Guidance Document No. 19 GUIDANCE ON SURFACE WATER CHEMICAL MONITORING UNDER THE WATER FRAMEWORK DIRECTIVE, Office for Official Publications of the European Communities, Luxembourg, 2009.
[47] V. Dulio, M. Carere, G. Hanke, S. Polesello, M. David, C. Sollazzo, et al., Guidance document No. 25 Guidance on chemical monitoring of sediment and biota under the Water Framework Directive, Office for Official Publications of the European Communities, Luxembourg, 2010. doi:10.2779/43586.
[48] W. Brack, V. Dulio, M. Ågerstrand, I. Allan, R. Altenburger, M. Brinkmann, et al., Towards the review of the European Union Water Framework management of chemical contamination in European surface water resources, Sci. Total Environ. 576 (2017) 720-737. doi:10.1016/j.scitotenv.2016.10.104.
[49] T. Rusina, F. Smedes, J. Klanova, Diffusion coefficients of polychlorinated biphenyls and polycyclic aromatic hydrocarbons in polydimethylsiloxane and low-density polyethylene polymers, J. Appl. Polym. Sci. 116 (2010) 1803-1810.
[50] B. Englert, Method 1694: Pharmaceuticals and Personal Care Products in Water, Soil, Sediment, and Biosolids by HPLC/MS/MS, Washington DC, 2007.
10 Protokol o odběru vzorků (vzor)
Označení vzorkovače: _________________________________________________________
|
Odběrová lokalita |
||
|
Číslo záznamníku teploty vody: |
ZAČÁTEK ODBĚRU Instalace vzorkovače |
KONEC ODBĚRU Deinstalace vzorkovače |
|
Odběrový technik |
||
|
GPS Zeměpisná šířka |
||
|
GPS Zeměpisná délka |
||
|
Datum a čas odběru |
||
|
Doba manipulace se vzorkovači (expozice terénní kontroly na vzduchu) |
||
|
Označení použitého terénního kontrolního vzorkovače |
||
|
Vodivost vody |
||
|
pH vody |
||
|
Teplota vody |
||
|
Teplota vzduchu |
||
|
Fotografický snímek - čas snímku |
||
11 Stanovení distribučních koeficientů sledovaných látek v soustavě adsorbent-voda (KSW)
11.1 Princip metody
Hodnota rozdělovacího koeficientu látky v systému vzorkovač-voda (KSW) kvantifikuje poměr koncentrace sledované látky v adsorbentu vzorkovače a její volně rozpuštěné koncentrace ve vodné fázi v rovnovážném stavu. Hodnoty KSW jsou potřebné pro přepočet koncentrace látek v pasivním vzorkovači na koncentraci ve vodné fáze. Stanovení se provádí v laboratorních podmínkách měřením koncentračního poměru látky v adsorbentu, a ve zvoleném objemu vody, do které byly přidány sledované látky, po ustálení distribuční rovnováhy sorbentu s vodní fází.
11.2 Stanovení KSW
Koncentrace sledovaných látek v extraktech vzorkovače a vodních roztocích byly převedeny na jednotky ng kg-1 (vzorkovač) a ng l-1 (vodní roztok). Hodnoty rozdělovacího koeficientu KSW byly vypočítány jako:

Měření bylo provedeno při 4 různých nominálních koncentracích sledovaných látek ve vodním roztoku (0.001-10 µg l-1), při pH=7 a teplotě 23°C. Vypočítané mediánové hodnoty log KSW pro různé testované adsorbenty jsou uvedeny v Tabulka 4 a 5. Experimentální hodnoty KSW se nachází i v souborech Distribucni_koeficienty_Ksw_PFC_latky.xlsx a Distribucni_koeficienty_Ksw_3farmaka.xlsx a tvoří jako samostatné přílohy součást této metodiky.
Tabulka 4. Hodnoty distribučního koeficientu perfluorovaných látek v systému adsorbent-vodní roztok (log KSW) při pH=7.0 a teplotě 23 °C. Hodnoty uvedené v tabulce jsou mediánové hodnoty měření provedených při 4 různých nominálních koncentracích sledovaných látek ve vodním roztoku (0.00110 µg l-1). Hodnoty pro adsrobent Oasis HLB® jsou zvýrazněny, protože tento sorbent byl vybrán pro konstrukci pasivních vzorkovačů uvedených v metodice.
|
Látka |
log Kow |
Oasis HLB |
Bakerbond Speedisk H2O-Philic DVB |
Amberlite XAD7 |
Oasis MAX |
Empore Disk anion exchange SR |
AlteSil |
|
PFPA |
3,43 |
2,94 |
2,60 |
1,39 |
4,87 |
3,13 |
0,07 |
|
PFHxA |
4,06 |
3,59 |
3,60 |
2,01 |
4,79 |
3,10 |
-0,13 |
|
PFHpA |
4,67 |
3,78 |
4,37 |
2,48 |
4,71 |
2,86 |
0,16 |
|
PFOA |
5,30 |
4,17 |
5,03 |
2,65 |
4,77 |
3,65 |
-0,10 |
|
PFDA |
6,50 |
4,59 |
5,13 |
3,10 |
4,82 |
3,08 |
0,05 |
|
PFDoDA |
7,77 |
4,50 |
5,10 |
3,48 |
4,52 |
3,13 |
1,33 |
|
PFTrDA |
8,25 |
4,35 |
5,14 |
3,64 |
4,30 |
3,08 |
2,06 |
|
PFBS |
3,90 |
3,66 |
3,76 |
2,30 |
4,26 |
3,25 |
-0,10 |
|
PFHxS |
5,17 |
4,25 |
5,15 |
2,77 |
4,29 |
2,90 |
-0,19 |
|
PFOS |
6,43 |
4,68 |
5,45 |
3,12 |
4,33 |
2,92 |
-0,22 |
|
FOSA |
5,62 |
4,91 |
5,71 |
3,50 |
5,16 |
2,65 |
2,43 |
|
PFNA |
5,92 |
4,40 |
5,16 |
2,85 |
4,77 |
3,29 |
0,05 |
Tabulka 5. Hodnoty distribučního koeficientu vybraných farmak v systému adsorbent-vodní roztok (log KSW) při pH=7.0 a teplotě 23 °C. Hodnoty uvedené v tabulce jsou mediánové hodnoty měření provedených při 3 různých nominálních koncentracích sledovaných látek ve vodním roztoku (0.1-10 µg l-1). Hodnoty pro adsorbent Oasis HLB® jsou zvýrazněny, protože tento sorbent byl vybrán pro konstrukci pasivních vzorkovačů uvedených v metodice.
|
Látka |
log Kow |
Oasis HLB |
Oasis MAX |
Empore Disk SDB RPS |
AlteSil |
XAD7 |
|
karbamazepin |
2,45 |
5,20 |
5,26 |
3,21 |
1,71 |
2,74 |
|
sulfamethoxazol |
0,89 |
4,40 |
5,13 |
2,89 |
-0,26 |
2,52 |
|
diklofenak |
4,51 |
4,37 |
5,27 |
3,04 |
1,53 |
2,03 |
12 Stanovení difůzních koeficientů perfluorovaných látek a vybraných farmak v agarozovém hydrogelu (Dp)
12.1 Princip metody
Hodnoty difúzního koeficientu Dp byly vypočítány z distribuce koncentrace látky mezi 6 vrstev agarozového hydrogelu po známé době difúze numerickým řešením podle 2. Fickova zákona.
12.2 Stanovení difúzních koeficientů látek v agarozovém hydrogelu
Hodnoty difúzního koeficientu sledovaných látek byly vypočítány z distribuce koncentrace látky mezi 6 vrstev agarozového hydrogelu po známé době difúze numerickým řešením podle 2. Fickova zákona podle postupu uvedeného v praci Rusina a kol. (2010) [48]. Výsledné naměřené hodnoty difúzního koeficientu perfluorovaných látek a vybraných farmak v agarozovém hydrogelu jsou uvedeny v tabulce (Tabulka 6).
12.3 Postup stanovení
Na skleněnou desku o rozměrech 10×10 cm bylo na sebe navrstveno šest plátků hydrogelu o velikosti 6×6×0,1 cm. Tyto pláty hydrogelu byly pokládány na sebe, přičemž každý plátek hydrogelu byl nejdříve zvážen na laboratorních vahách s přesností na 0,01 g. Spodní (tzv. donorový) plát hydrogelu byl před experimentem obohacen vodním roztokem směsi perfluorovaných látek a léčiv. Obohacení hydrogelů bylo uskutečněno namočením hydrogelů do roztoku směsi sledovaných látek o koncentraci 2 µg ml-1 po dobu 24 hodin. Takto připravené navrstvené pláty hydrogelu byly shora zakryty dalším sklem o rozměrech 10×10 cm (Obrázek 132) a umístěny do ploché plastové nádoby s víkem, která byla vyložena navlhčenou papírovou utěrkou, aby nedocházelo k vysychání plátů hydrogelu během expozice. Do nádoby byl uložen teplotní záznamník, který snímal v pětiminutovém intervalu teplotu a intenzitu světla. Celá nádoba byla obalena alobalem a probíhala inkubace při teplotě 22,0 ± 0,1°C nebo v lednici při teplotě 5,4 ± 0,7°C. Po zvolené době difúze byly plátky hydrogelu rozebrány a každý ze šesti plátků byl umístěn za účelem extrakce sledované látky z plátků do samostatné vialky o objemu 40 ml, ve které bylo 35 ml destilované vody. Před instrumentální analýzou probíhalo ředění vzorků methanolem podle požadavků analytické metody. Měření difúzních koeficientů se uskutečnilo po expozici 1, 2, 4 a 24 hodin. Každé měření probíhalo ve třech opakováních.
Obrázek 13. Schéma uspořádání gelů pro měření difúzních koeficientů.
Pro každé opakování v každém čase byly získány hodnoty difúzních koeficientů. Výslednou hodnotu tvoří průměr vážený obrácenou hodnotou rozptylu daného modelovým výpočtem [49]. Hodnoty pro časy 24 hodin nebyly zahrnuty, protože se systém dostal do rovnováhy, co znemožňuje přesný výpočet difúzního koeficientu.
Z vypočtených difúzních koeficientů byly taktéž stanoveny teoretické vzorkovací rychlosti pro terénní vzorkovač GG (rovnice 12). Tyto jsou uvedeny spolu s difúzními koeficientmi při obou sledovaných teplotách (Tabulka 6).
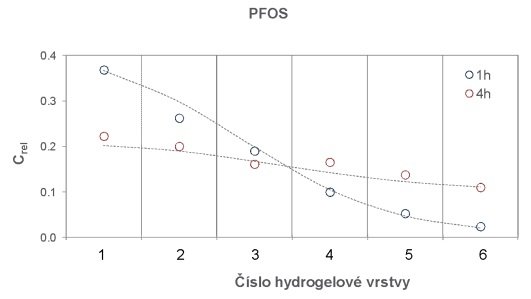
Obrázek 14 Distribuce PFOS v 6 naskládaných vrstvách 0,1 cm tlustých plátů agarozového hydrogelu po různé době inkubace při 22°C.
Výpočet váženého průměru difúzních koeficientů z dílčích měření:

D je vážený průměr difúzních koeficientů (cm2 s-1)
Di je difuzní koeficient z jednotlivého měření (cm2 s-1)
wi je váha pro vážený průměr
s2 je rozptyl stanovení při daných podmínkách
Výpočet teoretické vzorkovací rychlosti podle difúzních koeficientů:

RS je vzorkovací rychlost (ml d-1)
A je plocha vzorkovače (22,68 cm2)
D je difúzní koeficient látky při sledované teplotě (cm2 s-1)
d je tloušťka difúzní vrstvy hydrogelu (cm)
k je konstanta propočtu jednotek času (86400 s d-1)
Tabulka 6. Hodnoty difúzních koeficientů D a teoretických vzorkovacích rychlostí RS perfluorovaných látek. Při označení * byly koncentrace látek ve vrstvě gelu pod limitem kvantifikace.
|
Látka |
D (22°C) ×106 cm2 s-1 |
D (5°C) ×106 cm2 s-1 |
RS (22°C) ml d-1 |
RS (5°C) ml d-1 |
|
PFPA |
4,79 |
4,03 |
93,9 |
78,9 |
|
PFHxA |
4,52 |
3,74 |
88,6 |
73,4 |
|
PFHpA |
4,81 |
3,44 |
94,2 |
67,5 |
|
PFOA |
4,38 |
3,39 |
85,9 |
66,4 |
|
PFNA |
3,51 |
3,27 |
68,9 |
64,1 |
|
PFDA |
3,38 |
1,87 |
66,3 |
36,7 |
|
PFDoDA |
1,37 |
* |
26,9 |
* |
|
PFTRDA |
4,84 |
* |
9,5 |
* |
|
PFHxS |
4,58 |
4,42 |
89,7 |
86,6 |
|
PFBS |
4,98 |
4,95 |
97,6 |
96,9 |
|
PFOS |
4,09 |
3,48 |
80,2 |
68,2 |
|
FOSA |
1,13 |
2,27 |
22,1 |
44,4 |
Tabulka 7. Hodnoty difúzních koeficientů D a teoretických vzorkovacích rychlostí RS vybraných farmak. Při označení byly koncentrace látek ve vrstvě gelu pod limitem kvantifikace.
|
Látka |
D (22°C) ×106 cm2 s-1 |
D (5°C) ×106 cm2 s-1 |
RS (22°C) ml d-1 |
RS (5°C) ml d-1 |
|
Karbamazepin |
6,77 |
3,67 |
132,7 |
72,0 |
|
Diklofenak |
3,64 |
3,18 |
71,2 |
62,3 |
|
Sulfamethoxazol |
3,93 |
3,58 |
77,1 |
70,2 |
Experimentální hodnoty difúzních koeficientů D v agarozovém hydrogelu se nachází i v souboru Difuzni_koeficienty.xlsx a tvoří jako samostatná příloha součást této metodiky.
13 Kalibrace pasivního vzorkovače v průtokovém systému
Při kalibraci vzorkovačů v laboratorních podmínkách jsou vzorkovače exponovány v průtokovém systému s kontrolovanou teplotou vody, turbulencí a koncentrací sledovaných analytů ve vodě.
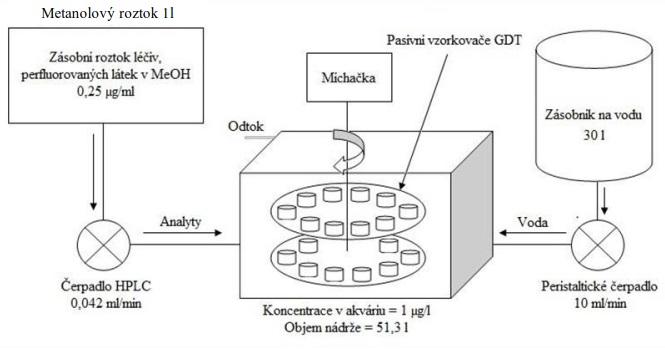
Obrázek 15 Schéma kalibrace pasivních vzorkovačů v průtokovém systému.
Pasivní vzorkovače byly během expozice v roztoku analytů v nádrži připevněny na otáčivou konstrukci, která byla hřídelem napojena na laboratorní míchačku. HPLC čerpadlo čerpalo do nádrže směsný roztok analytů o koncentraci 0,25 µg ml-1. Tento roztok byl připraven ze zásobních roztoků analytů zředěním do methanolu. Voda byla do systému čerpána ze zásobníku o objemu 30 litrů peristaltickým čerpadlem. Koncentrace látek v nádrži se udržovala na hodnotě přibližně 1 µg l-1 díky konstantímu proudění methanolového roztoku a vody do systému a odtoku ze systému, aby v důsledku sorpce látek do vzorkovačů, materiálů zařízení nebo rozkladu nedošlo k poklesu koncentrace (Obrázek 4).
Rychlost otáčení kovové konstrukce byla nastavena na 36 otáček za minutu. Odběry vzorkovačů probíhaly po 34, 70, 106, 142, 178, 214, 250 a 286 hodinách. Na odebrané vzorkovače byla Pasteurovou pipetou přidána kapka azidu sodného pro zabránění růstu mikroorganismů a vzorkovače byly umístěny do uzavíratelných plastových „zip lock“ sáčků s malým množstvím destilované vody. Na místa odebraných vzorkovačů byla doplněna prázdná pouzdra pasivních vzorkovačů, která sloužila k zachování konstantních hydrodynamických podmínek. Pro zjištění koncentrace látek v roztoku v nádrži bylo při každém odběru pasivních vzorkovačů odebráno také 3×100 ml vody z nádrže, která pak byla zpracována metodou SPE.
Byly použity kolonky Oasis HLB® 60 mg. Kondicionovány byly pomocí 3 ml methanolu a 3 ml vody okyselené kyselinou chlorovodíkovou na pH 2 - 3. Pak byl převeden vzorek, rovněž okyselen na pH 2-3. Kolonky byly promyty okyselenou destilovanou vodou. Analyty byly eluovány pomocí 3 ml methanolu. Vzorky byly odpařeny pod proudem dusíku do objemu 0,5 ml a naředěny vodou v poměru dle použité analytické metody.
Vzorkovací rychlosti RS (ml d-1) u perflurovaných látek byly vypočítány ze vztahu

kde β0 (ng g-1 d-1) je směrnice lineárního nárůstu koncentrace sledované látky v čase, mS (g) je hmotnost sorbentu ve vzorkovači, a cW (ng ml-1) je průměrná koncentrace látky ve vodě během kalibrace vzorkovačů. Naměřené rychlosti pak byly propočítány pro vzorkovače použité v terénu (RST) podle poměru vzorkovacích ploch.

Tabulka 8. Vzorkovací rychlosti perfluorovaných látek a vybraných farmak pro pasivní vzorkovače s difúzní vrstvou hydrogelu a plochou 3,14 cm2 (Rs, z laboratorní kalibrace) a 22,68 cm2 (aplikováno při použití vzorkovačů v terénu).
|
Látka |
RSlab ml d-1 |
RSterén ml d-1 |
|
PFPA |
21 |
149 |
|
PFHxA |
10 |
74 |
|
PFHpA |
18 |
128 |
|
PFOA |
14 |
100 |
|
PFNA |
14 |
100 |
|
PFDA |
10 |
74 |
|
PFBS |
10 |
75 |
|
PFHxS |
11 |
78 |
|
PFOS |
10 |
70 |
|
FOSA |
8 |
60 |
|
Diklofenak |
13 |
94 |
|
Karbamazepin |
19 |
139 |
|
Sulfametoxazol |
10 |
70 |
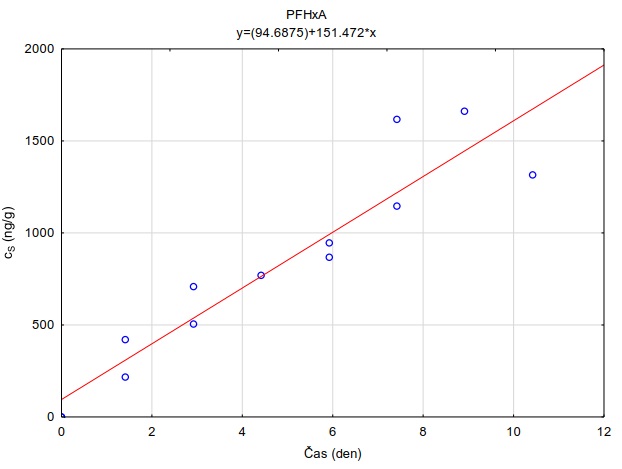
Obrázek 16. Kalibrační závislost koncentrace PFHxA na jednotku hmotnosti sorbentu v čase.
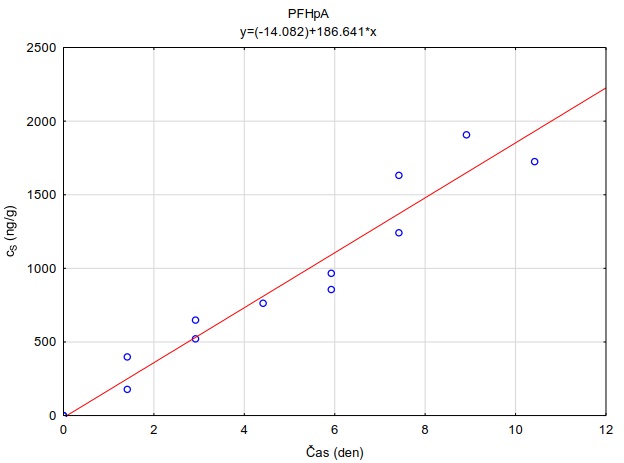
Obrázek 17. Kalibrační závislost koncentrace PFHpA na jednotku hmotnosti sorbentu v čase.
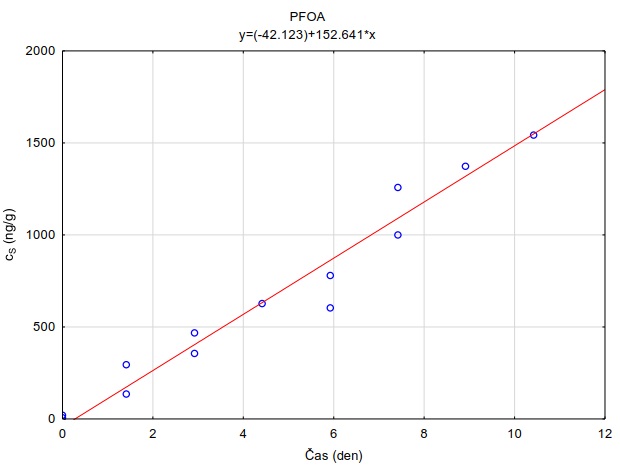
Obrázek 18. Kalibrační závislost koncentrace PFOA na jednotku hmotnosti sorbentu v čase.
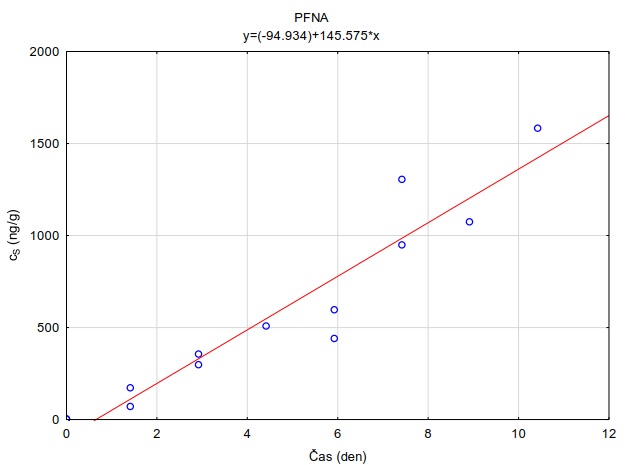
Obrázek 19. Kalibrační závislost koncentrace PFNA na jednotku hmotnosti sorbentu v čase.
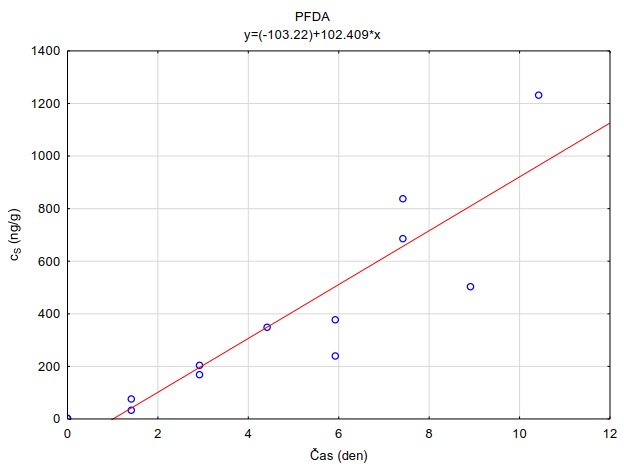
Obrázek 20. Kalibrační závislost koncentrace PFDA na jednotku hmotnosti sorbentu v čase.
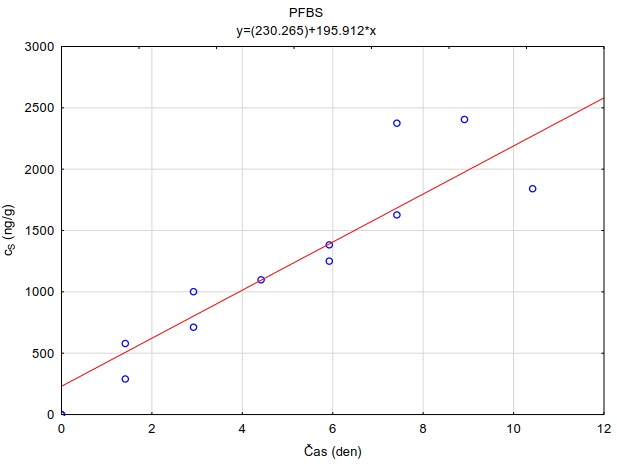
Obrázek 21. Kalibrační závislost koncentrace PFBS na jednotku hmotnosti sorbentu v čase.
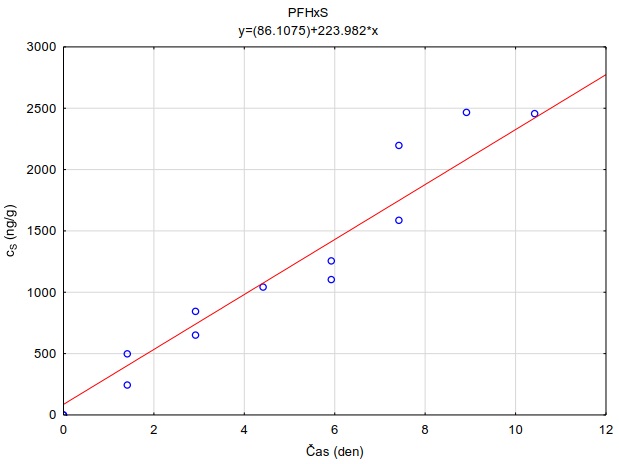
Obrázek 22. Kalibrační závislost koncentrace PFHxS na jednotku hmotnosti sorbentu v čase.
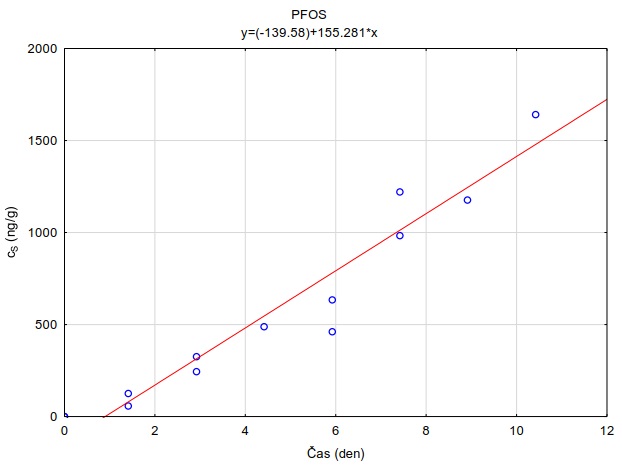
Obrázek 23. Kalibrační závislost koncentrace PFOS na jednotku hmotnosti sorbentu v čase.
Obrázek 24. Kalibrační závislost koncentrace FOSA na jednotku hmotnosti sorbentu v čase.
Obrázek 25. Kalibrační závislost koncentrace diklofenaku na jednotku hmotnosti sorbentu v čase.
Obrázek 26. Kalibrační závislost koncentrace karabamazepinu na jednotku hmotnosti sorbentu v čase.
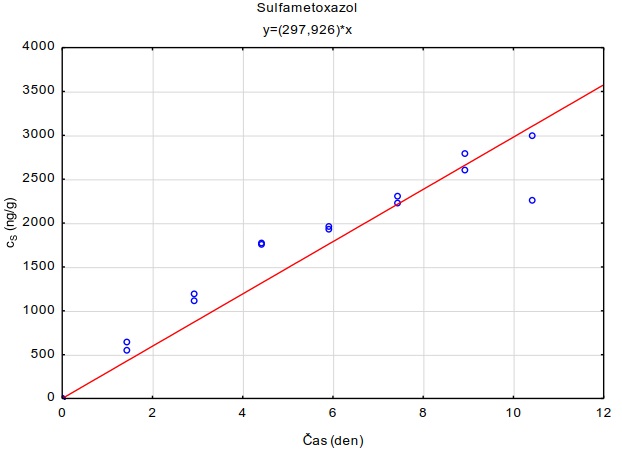
Obrázek 27. Kalibrační závislost koncentrace sulfomethoxazolu na jednotku hmotnosti sorbentu v čase.
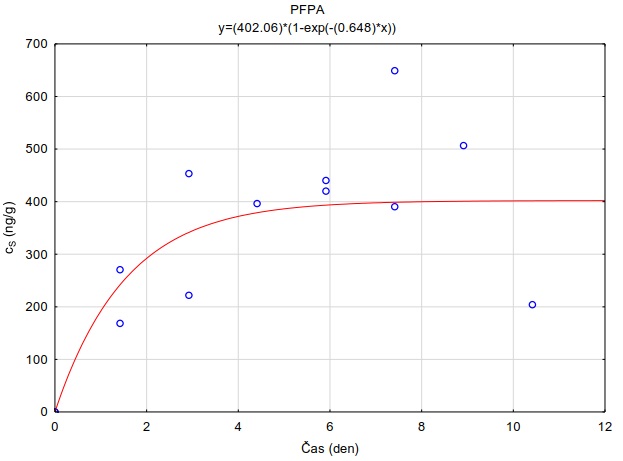
Obrázek 28. Kalibrační závislost koncentrace PFPA na jednotku hmotnosti sorbentu v čase.
14 Příklady podmínek instrumentální analýzy
14.1 Perfluorované látky
Pro separaci analyzovaných látek je využívána krátká kolona (do 50 mm) s malými částicemi nepolární stacionární fáze, nejčastěji C18 do velikosti 4 µm, která umožňuje separaci cílových analytů v krátkém čase. Pro kvantifikaci je nutné využívat směs značených injekčních standardů, které dovolí postižení případných změn v ionizaci jednotlivých analytů vlivem přítomné matrice vzorku. K separaci polárních perfluorovaných látek je obvykle využívána gradientová eluce dvou složek mobilní fáze (vody a methanolu, nejčastěji s přídavkem octanu amonného pro zvýšení ionizace analytů v iontovém zdroji hmotnostního spektrometru. Obě složky mobilní fáze se před použitím vloží na 20 min do ultrazvukové lázně k odvzdušnění. Před každou sadou vzorků je promyto čerpadlo kapalinového chromatografu, a to nejprve čistým methanolem po dobu nejméně 5 minut o maximálním průtoku a poté druhou mobilní složkou o stejném průtoku a po stejnou dobu. Dále je kolona promyta adekvátním průtokem methanolu po dobu 20 minut a poté mobilní fází stejným průtokem po stejnou dobu. Takto kondicionovaná kolona je připravena k použití. Před samotným měřením každé sady vzorků jsou proměřeny roztoky kalibračních standardů a porovná se stabilita retenčních časů, odezev a rozlišení. Poté se každých 8-10 vzorků měří kalibrační standard pro ověření stability během měření reálných vzorků. Hmotnostní spektrometr je pro maximální výtěžnost ionozace sledovaných perfluorovaných látek nastaven na negativní mód. Příklad nastavení parametrů hmotnostního detektoru pro splnění výše uvedeného je v Tabulkách 9 a 10.
Tabulka 9. Parametry nastavení iontového zdroje hmotnostního spektrometru
|
ESI mód |
negativní |
|
napětí iontového zdroje |
-4500 V |
|
teplota iontového zdroje |
450°C |
|
tlak zmlžujícího plynu |
345 kPa |
|
tlak sušícího plynu |
207 kPa |
Tabulka 10. Parametry nastavení hmotnostního spektrometru
|
Název látky |
Prekurzorový ion (Da) |
Produktový ion (Da) |
Kolisní energie (eV) |
|
PFPA |
263 |
219, 241 |
-10, -5 |
|
PFHxA |
313 |
269, 219 |
-13, -26 |
|
PFHpA |
363 |
319, 169 |
-15, -24 |
|
PFOA |
413 |
369, 169 |
-17, -24 |
|
PFNA |
463 |
419, 219 |
-15, -24 |
|
PFDA |
513 |
469, 219 |
-18, -26 |
|
PFDoDA |
613 |
569, 319 |
-22, -26 |
|
PFTrDA |
663 |
619, 169 |
-19, -36 |
|
PFBS |
299 |
99, 80 |
-70, -40 |
|
PFHxS |
399 |
99, 80 |
-60, -45 |
|
PFOS |
499 |
99, 80 |
-120, -90 |
|
FOSA |
498 |
78, 64 |
-84, -130 |
Pro výpočet koncentrace sledovaných látek ve vzorcích je využívána interní kalibrace s využitím značeného vnitřního standardu. Ke konstrukci kalibrační křivky se na osu y vynáší poměr plochy píku analytu a plochy injekčního standardu a na osu x poměr koncentrace analytu a injekčního standardu. Pro jednotlivé analyty jsou jako injekční standardy použity příslušné izotopicky značené látky, nebo standardy izotopicky značených látek s podobnou délkou alifatického řetězce v příslušné skupině. Pro konstrukci kalibračních křivek může být využit volně dostupný software pro statistické výpočty, stejně jako speciální software dodaný výrobcem hmotnostního spektrometru. Nejčastěji se konstruují kalibrační s využitím lineárních rovnic v celém rozsahu. Standardům, které se nacházejí v oblasti nízkých koncentrací je obvykle přiřazována vyšší váha (tzv. 1/x vážení).
Vzhledem k závislosti limitů kvantifikace na použité matrici, jsou zde uváděny pouze typické instrumentální detekční limity (IDL), které jsou dosažitelné stávající instrumentální technikou z kalibračních křivek čistých roztoků.
Tabulka 11. Typické instrumentální detekční limity metody
|
Název látky |
IDL (pg ml-1) |
|
PFPA |
4 |
|
PFHxA |
10 |
|
PFHpA |
13 |
|
PFOA |
28 |
|
PFNA |
27 |
|
PFDA |
14 |
|
PFUnDA |
103 |
|
PFDoDA |
25 |
|
PFTrDA |
15 |
|
PFBS |
40 |
|
PFHxS |
17 |
|
PFOS |
10 |
|
FOSA |
4 |
14.2 Farmaka
Léčiva jsou separována a analyzována s použitím kapalinové chromatografie ve spojení s hmotností detekcí. Stanovované sloučeniny jsou dle metody EPA 1694 [50] a na základě jejich chování při separaci a detekci, rozděleny do dvou skupin. Pro separaci obou skupin je vhodné využít kolona do 1000 mm s malými částicemi nepolární stacionární fáze, nejčastěji C18 do velikosti 3,5 µm. Pokud je při separaci využito menších částic stacionární fáze, je nutné pracovat s kapalinovým chromatografem vybaveným čerpadlem umožňujícím pracovat při vysokých tlacích, tzv. ultra účinným kapalinovým chromatografem. Pro separaci obou skupin léčiv se jako složky mobilní fáze se používá nejčastěji methanol, nebo acetonitril v kombinaci s vodou, jako mobilní fází. Do obou složek mobilní fáze je možné přidat modifikátory pro zlepšení ionizace cílových analytů v ionotovém zdroji, jako například kyselina mravenčí a octová, popř. octan amonný. Pro separaci látek je pak využita gradientová eluce složek mobilní fáze tak, aby došlo k oddělení cílových analytů. Pro kondicionaci pumpy a kolony je využíván stejný postup, který je popsaný u instrumentální analýzy perfluorovaných látek.
Hmotnostní spektrometr je pro maximální výtěžnost ionizace cílových polutatnů nastaven buď na positivní, nebo negativní mód. Napětí a teplota, stejně jako další podmínky v iontovém zdroji jsou nastaveny tak, aby bylo dosaženo instrumentálních limitů. Typické instrumentální limity detekce jsou 1,1 pg ml-1 pro diklofenak, 1,7 pg ml-1 pro sulfomethoxazol a 0,07 pg ml-1 pro karbamazepin. Příklad podmínek hmotnostního detektoru pro splnění výše uvedeného jsou následující: kapilární napětí v iontovém zdroji 2,5 kV - 2,5 kV; desolvatační teplota 350°C a průtok desolvatačního plynu (dusíku) 700 l hod-1.
Pro kvalitativní určení analyzovaných léčiv jsou detekovány 2 produktové ionty vzniklé z mateřského prekurzorového iontu, tzv. MRM (multiple reaction monitoring) přechody, které jsou uvedené v tabulce 12. Intenzita optimalizovaného prvního MRM přechodu sloužila pro výpočet přesné koncentrace látek ve vzorcích (kvantifikace), druhý pro potvrzení přítomnosti detekované látky (konfirmace). Pro kvantitativní analýzu byly změřeny kalibrační závislosti v koncentračním rozsahu 0,005 -100 ng ml-1.
Tabulka 12. Parametry nastavení hmotnostního spektrometru
|
Název látky |
Prekurzorový ion (Da) |
Produktový ion (Da) |
Kolisní energie (eV) |
|
|
Karbamazepin |
ESI+ |
237 |
194; 179 |
20; 35 |
|
Diklofenak |
ESI+ |
296 |
214 |
16 |
|
Sulfamethoxazol |
ESI+ |
254 |
156; 92 |
16; 26 |
1 V některých případech se však výsledky mohou lišit, protože změny koncentrace probíhající v době mezi dvěma odběry, bodový odběr nepostřehne, avšak pasivním vzorkování ano, což je jedna z hlavních výhod metody pasivního vzorkování.